

Drug treatment of pulmonary hypertension in children
- PMID: 24114695
- PMCID: PMC3946901
- DOI: 10.1007/s40272-013-0052-2
Drug treatment of pulmonary hypertension in children
Abstract
Pulmonary arterial hypertension (PAH) is a rare disease in infants and children that is associated with significant morbidity and mortality. The disease is characterized by progressive pulmonary vascular functional and structural changes resulting in increased pulmonary vascular resistance and eventual right heart failure and death. In the majority of pediatric patients, PAH is idiopathic or associated with congenital heart disease and rarely is associated with other conditions such as connective tissue or thromboembolic disease. Although treatment of the underlying disease and reversal of advanced structural changes has not yet been achieved with current therapy, quality of life and survival have been improved significantly. Targeted pulmonary vasodilator therapies, including endothelin receptor antagonists, prostacyclin analogs, and phosphodiesterase type 5 inhibitors, have demonstrated hemodynamic and functional improvement in children. The management of pediatric PAH remains challenging, as treatment decisions continue to depend largely on results from evidence-based adult studies and the clinical experience of pediatric experts. This article reviews the current drug therapies and their use in the management of PAH in children.
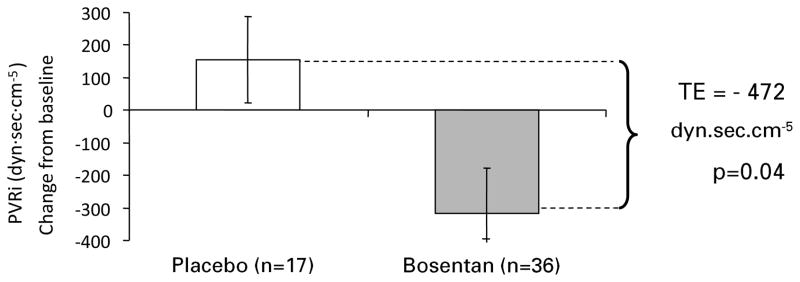
Figures


















References
-
- Rubin LJ. Primary pulmonary hypertension. N Engl J Med. 1997;336(2):111–7. - PubMed
-
- Allen KM, Haworth SG. Cytoskeletal features of immature pulmonary vascular smooth muscle cells: the influence of pulmonary hypertension on normal development. J Pathol. 1989 Aug;158(4):311–7. - PubMed
-
- Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004 Jun 16;43(12 Suppl S):13S–24S. - PubMed
-
- Mandegar M, Fung YC, Huang W, Remillard CV, Rubin LJ, Yuan JX. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc Res. 2004 Sep;68(2):75–103. - PubMed
-
- Hassoun PM, Mouthon L, Barbera JA, Eddahibi S, Flores SC, Grimminger F, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009 Jun 30;54(1 Suppl):S10–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
