

The somatic genomic landscape of glioblastoma
- PMID: 24120142
- PMCID: PMC3910500
- DOI: 10.1016/j.cell.2013.09.034
The somatic genomic landscape of glioblastoma
Erratum in
- Cell. 2014 Apr 24;157(3):753
Abstract
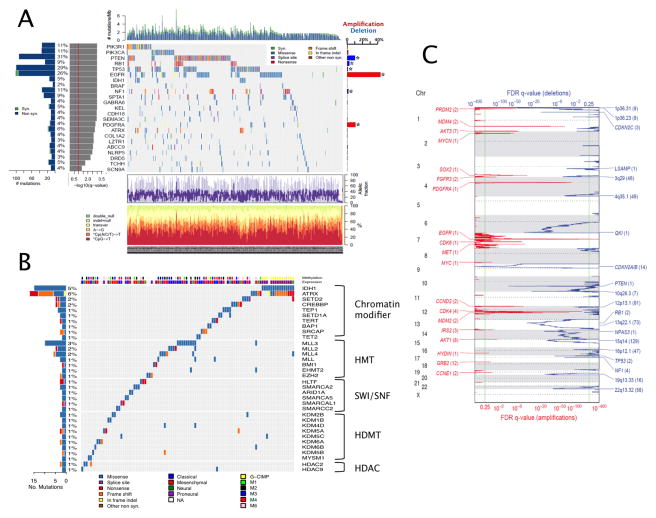
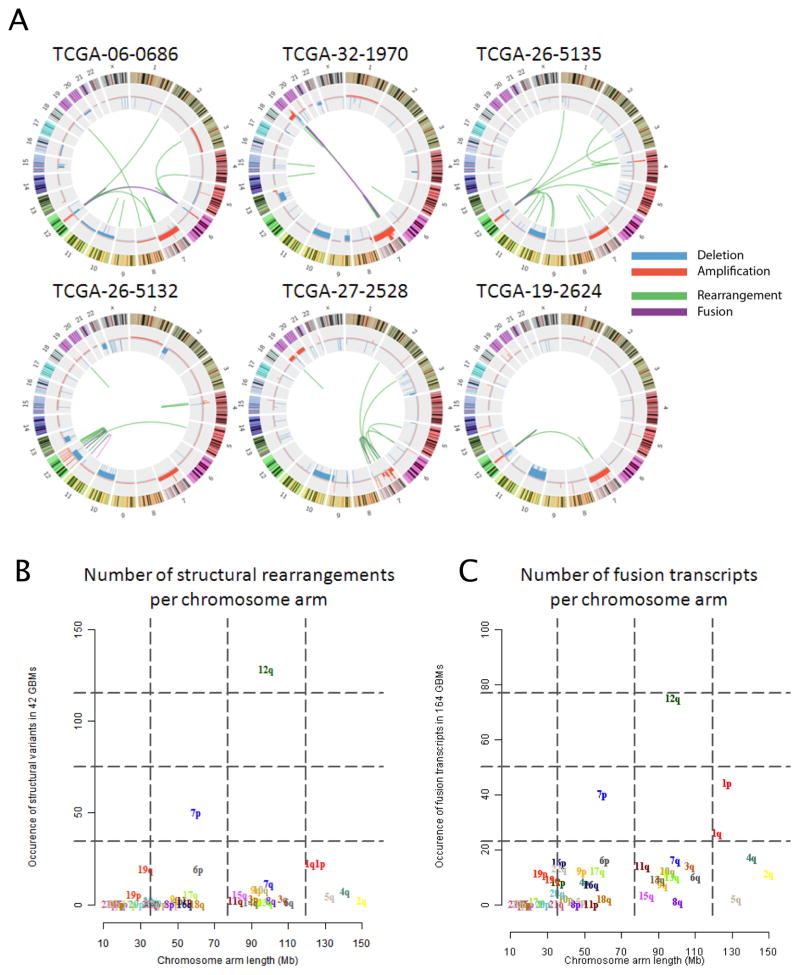
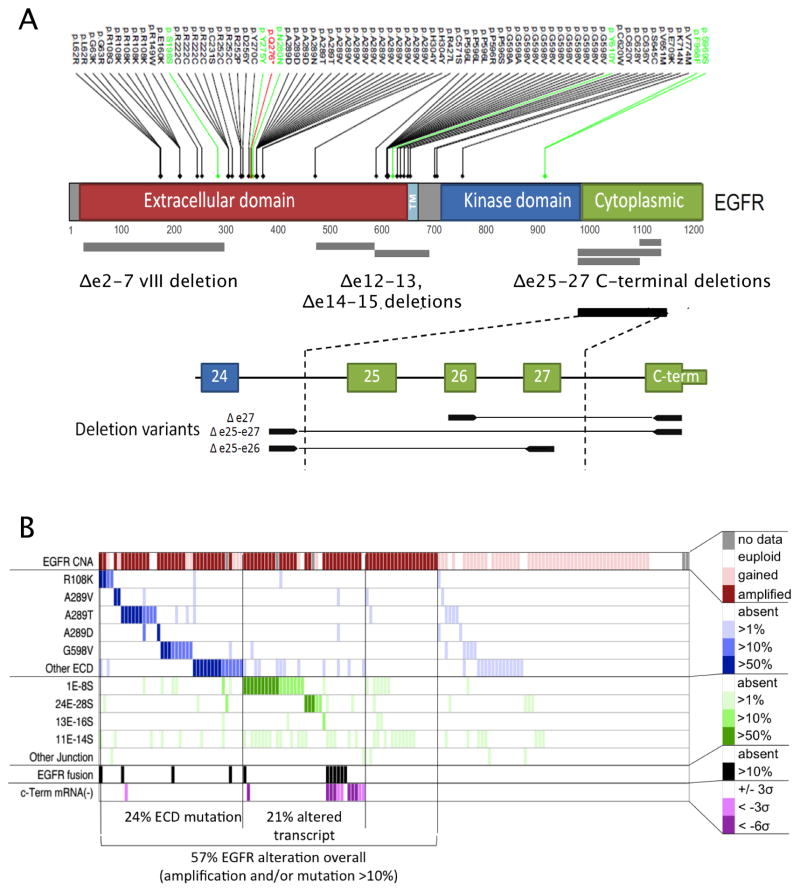
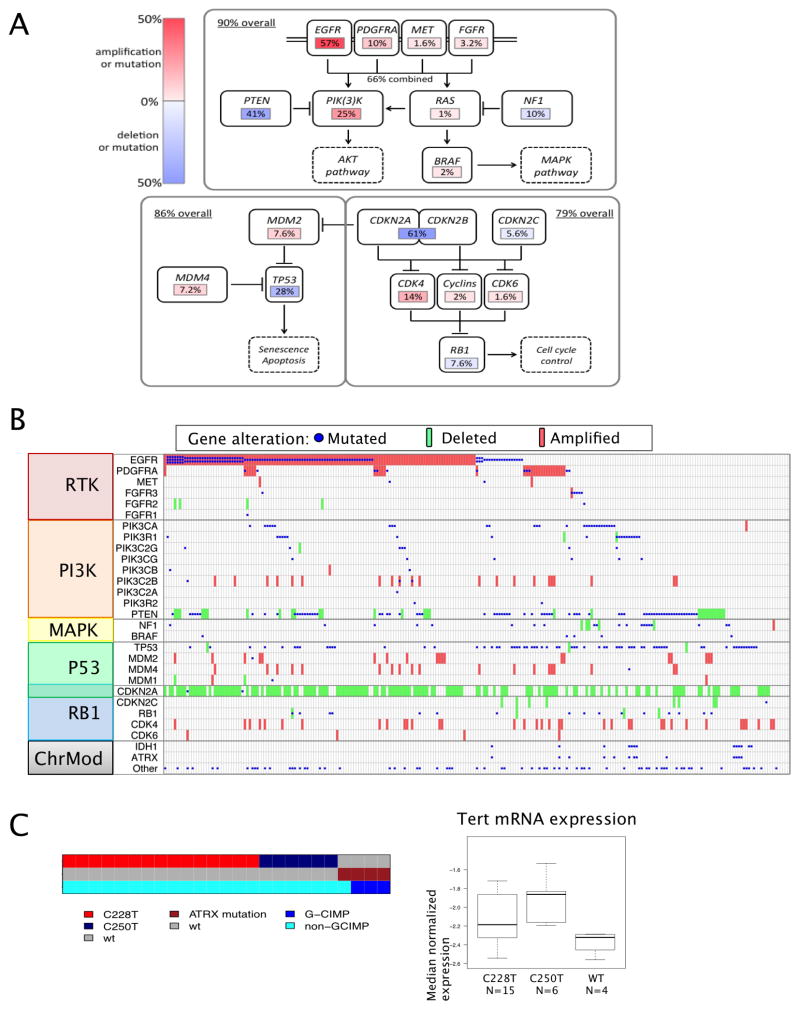
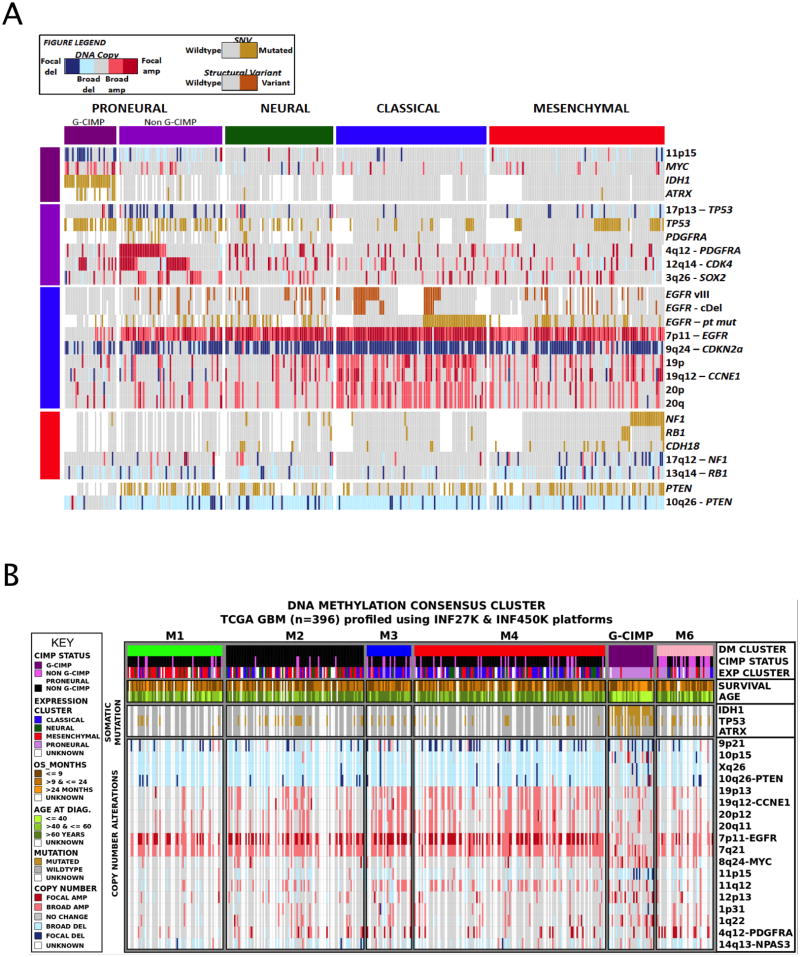
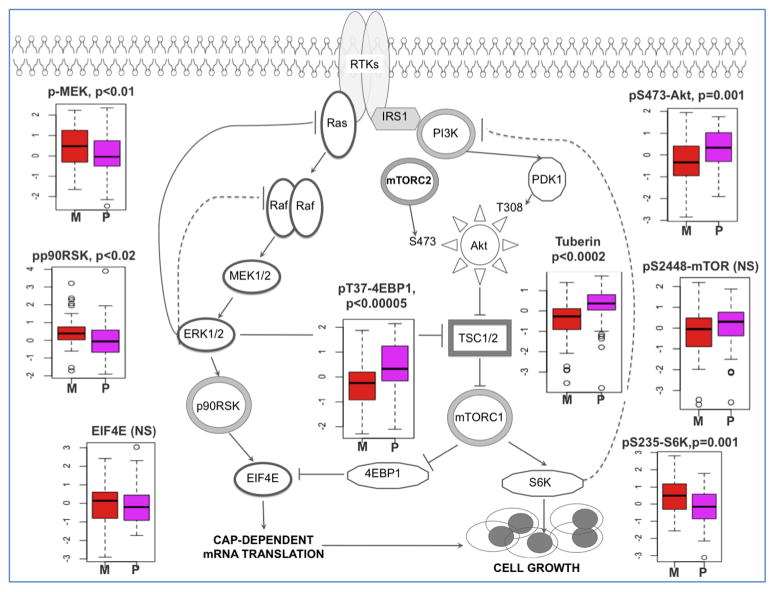
We describe the landscape of somatic genomic alterations based on multidimensional and comprehensive characterization of more than 500 glioblastoma tumors (GBMs). We identify several novel mutated genes as well as complex rearrangements of signature receptors, including EGFR and PDGFRA. TERT promoter mutations are shown to correlate with elevated mRNA expression, supporting a role in telomerase reactivation. Correlative analyses confirm that the survival advantage of the proneural subtype is conferred by the G-CIMP phenotype, and MGMT DNA methylation may be a predictive biomarker for treatment response only in classical subtype GBM. Integrative analysis of genomic and proteomic profiles challenges the notion of therapeutic inhibition of a pathway as an alternative to inhibition of the target itself. These data will facilitate the discovery of therapeutic and diagnostic target candidates, the validation of research and clinical observations and the generation of unanticipated hypotheses that can advance our molecular understanding of this lethal cancer.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
References
-
- Bady P, Sciuscio D, Diserens AC, Bloch J, van den Bent MJ, Marosi C, Dietrich PY, Weller M, Mariani L, Heppner FL, et al. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta neuropathologica. 2012;124:547–560. - PMC - PubMed
-
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U24CA143840/CA/NCI NIH HHS/United States
- U24 CA143882/CA/NCI NIH HHS/United States
- U24CA143731/CA/NCI NIH HHS/United States
- U24CA126551/CA/NCI NIH HHS/United States
- U24 CA143866/CA/NCI NIH HHS/United States
- U24 CA143843/CA/NCI NIH HHS/United States
- U24CA143867/CA/NCI NIH HHS/United States
- U24CA143799/CA/NCI NIH HHS/United States
- U24 CA143883/CA/NCI NIH HHS/United States
- R01 CA163722/CA/NCI NIH HHS/United States
- U24 CA143867/CA/NCI NIH HHS/United States
- U24 CA126546/CA/NCI NIH HHS/United States
- U24CA126561/CA/NCI NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U24 CA143835/CA/NCI NIH HHS/United States
- P50 CA108961/CA/NCI NIH HHS/United States
- U54HG003273/HG/NHGRI NIH HHS/United States
- U24CA126543/CA/NCI NIH HHS/United States
- U24CA126554/CA/NCI NIH HHS/United States
- U24CA143858/CA/NCI NIH HHS/United States
- U24CA126544/CA/NCI NIH HHS/United States
- U24CA143882/CA/NCI NIH HHS/United States
- U24 CA126551/CA/NCI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- U24CA144025/CA/NCI NIH HHS/United States
- U54HG003067/HG/NHGRI NIH HHS/United States
- U24 CA143845/CA/NCI NIH HHS/United States
- U24 CA143799/CA/NCI NIH HHS/United States
- P50 CA098258/CA/NCI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- U24 CA144025/CA/NCI NIH HHS/United States
- U24CA143883/CA/NCI NIH HHS/United States
- U54HG003079/HG/NHGRI NIH HHS/United States
- U24 CA126554/CA/NCI NIH HHS/United States
- U24 CA143840/CA/NCI NIH HHS/United States
- K08 NS045077/NS/NINDS NIH HHS/United States
- U24CA143835/CA/NCI NIH HHS/United States
- U24 CA126561/CA/NCI NIH HHS/United States
- U24 CA143858/CA/NCI NIH HHS/United States
- U24CA143843/CA/NCI NIH HHS/United States
- P30 CA015083/CA/NCI NIH HHS/United States
- U24 CA143848/CA/NCI NIH HHS/United States
- U24 CA126543/CA/NCI NIH HHS/United States
- U24CA143848/CA/NCI NIH HHS/United States
- U24CA126563/CA/NCI NIH HHS/United States
- U24CA126546/CA/NCI NIH HHS/United States
- U54 HG003079/HG/NHGRI NIH HHS/United States
- U24CA143866/CA/NCI NIH HHS/United States
- U24CA143845/CA/NCI NIH HHS/United States
- U24 CA126563/CA/NCI NIH HHS/United States
- U24 CA126544/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
