

Coupling heme and iron metabolism via ferritin H chain
- PMID: 24124891
- PMCID: PMC3961798
- DOI: 10.1089/ars.2013.5666
Coupling heme and iron metabolism via ferritin H chain
Abstract
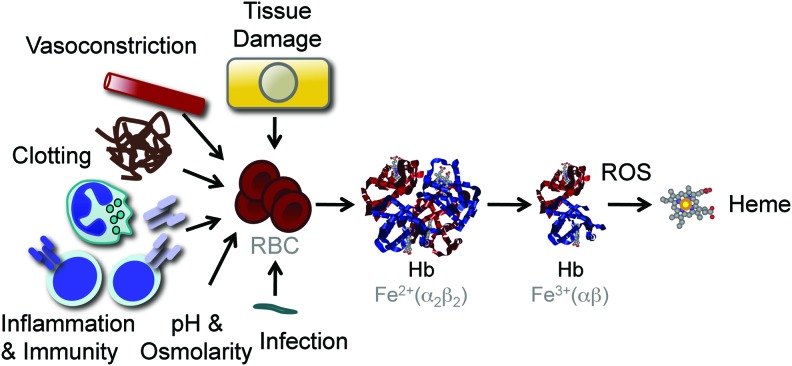
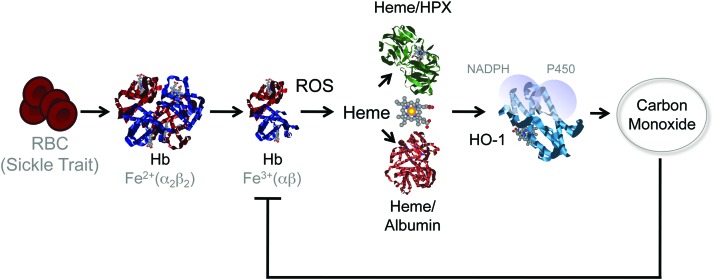
Significance: Inflammation and immunity can be associated with varying degrees of heme release from hemoproteins, eventually leading to cellular and tissue iron (Fe) overload, oxidative stress, and tissue damage. Presumably, these deleterious effects contribute to the pathogenesis of systemic infections.
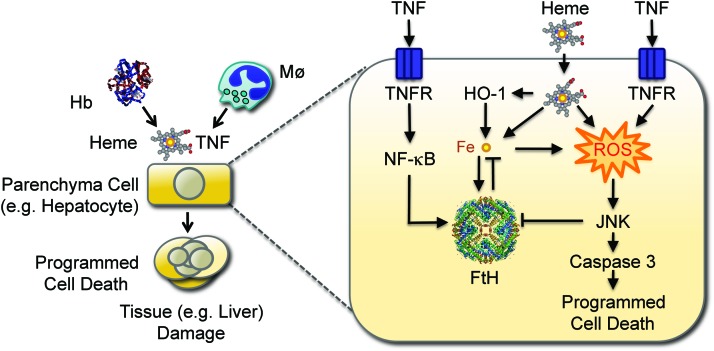
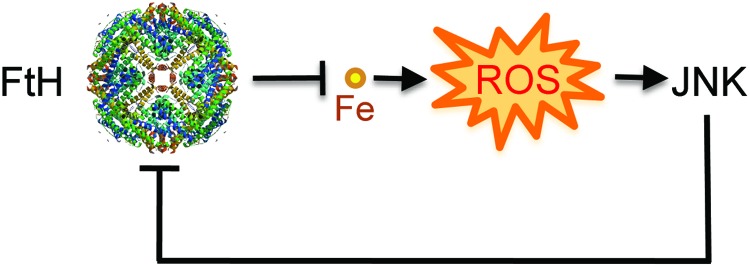
Recent advances: Heme release from hemoglobin sensitizes parenchyma cells to undergo programmed cell death in response to proinflammatory cytokines, such as tumor necrosis factor. This cytotoxic effect is driven by a mechanism involving intracellular accumulation of free radicals, which sustain the activation of the c-Jun N-terminal kinase (JNK) signaling transduction pathway. While heme catabolism by heme oxygenase-1 (HO-1) prevents programmed cell death, this cytoprotective effect requires the co-expression of ferritin H (heart/heavy) chain (FTH), which controls the pro-oxidant effect of labile Fe released from the protoporphyrin IX ring of heme. This antioxidant effect of FTH restrains JNK activation, whereas JNK activation inhibits FTH expression, a cross talk that controls metabolic adaptation to cellular Fe overload associated with systemic infections.
Critical issues and future directions: Identification and characterization of the mechanisms via which FTH provides metabolic adaptation to tissue Fe overload should provide valuable information to our current understanding of the pathogenesis of systemic infections as well as other immune-mediated inflammatory diseases.
Figures
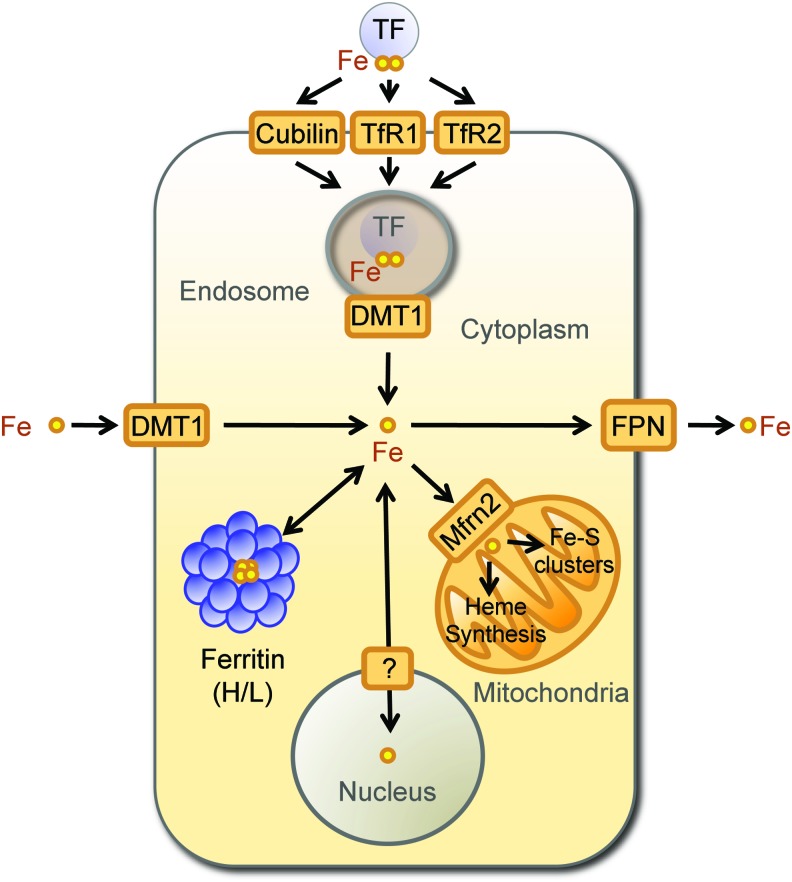
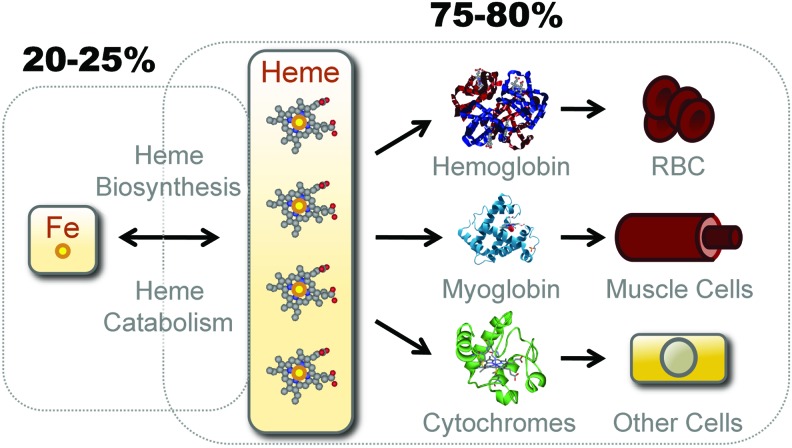
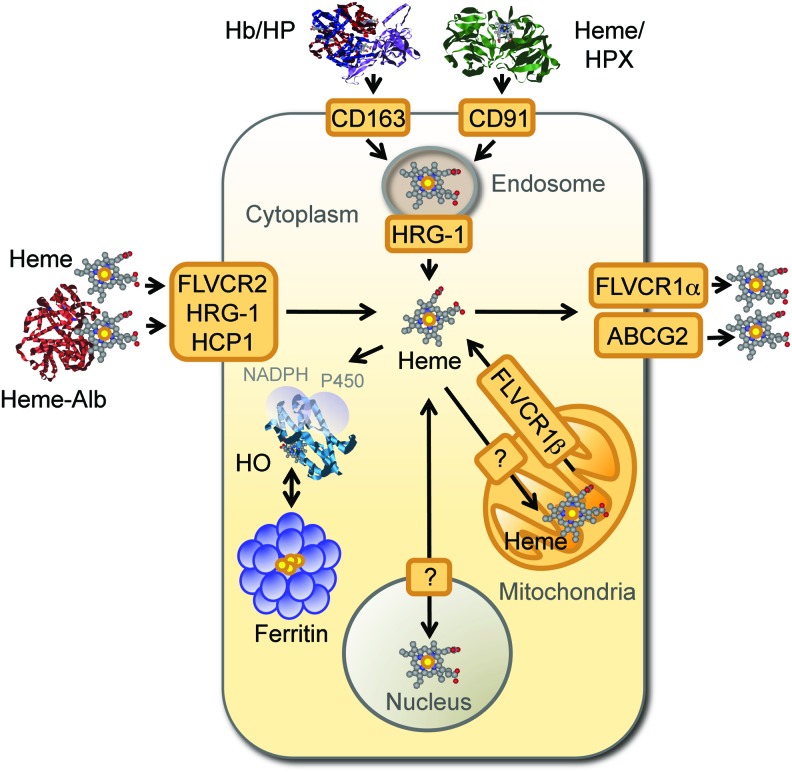
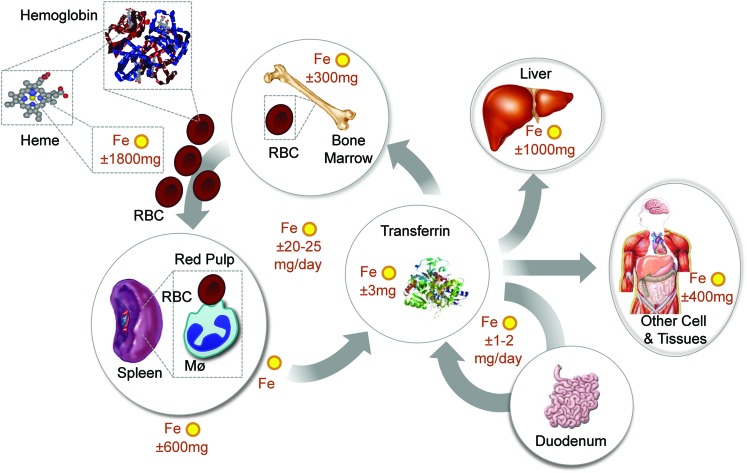
References
-
- Aisen P, Leibman A, and Zweier J. Stoichiometric and site characteristics of the binding of iron to human transferrin. J Biol Chem 253: 1930–1937, 1978 - PubMed
-
- Ajioka RS, Phillips JD, and Kushner JP. Biosynthesis of heme in mammals. Biochim Biophys Acta 1763: 723–736, 2006 - PubMed
-
- Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, and Cook JL. NRF2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem 274: 26071–26078, 1999 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
