

Near-identical segregation of mtDNA heteroplasmy in blood, muscle, urinary epithelium, and hair follicles in twins with optic atrophy, ptosis, and intractable epilepsy
- PMID: 24126373
- PMCID: PMC6551219
- DOI: 10.1001/jamaneurol.2013.4111
Near-identical segregation of mtDNA heteroplasmy in blood, muscle, urinary epithelium, and hair follicles in twins with optic atrophy, ptosis, and intractable epilepsy
Abstract
Importance: Mitochondrial DNA (mtDNA) disorders have emerged as major causes of inherited neurologic disease. Despite being well recognized for more than 2 decades, the clinical presentation continues to broaden. The phenotypic heterogeneity is partly owing to different percentage levels of mutant mtDNA heteroplasmy in different tissues, but the factors influencing this are poorly understood.
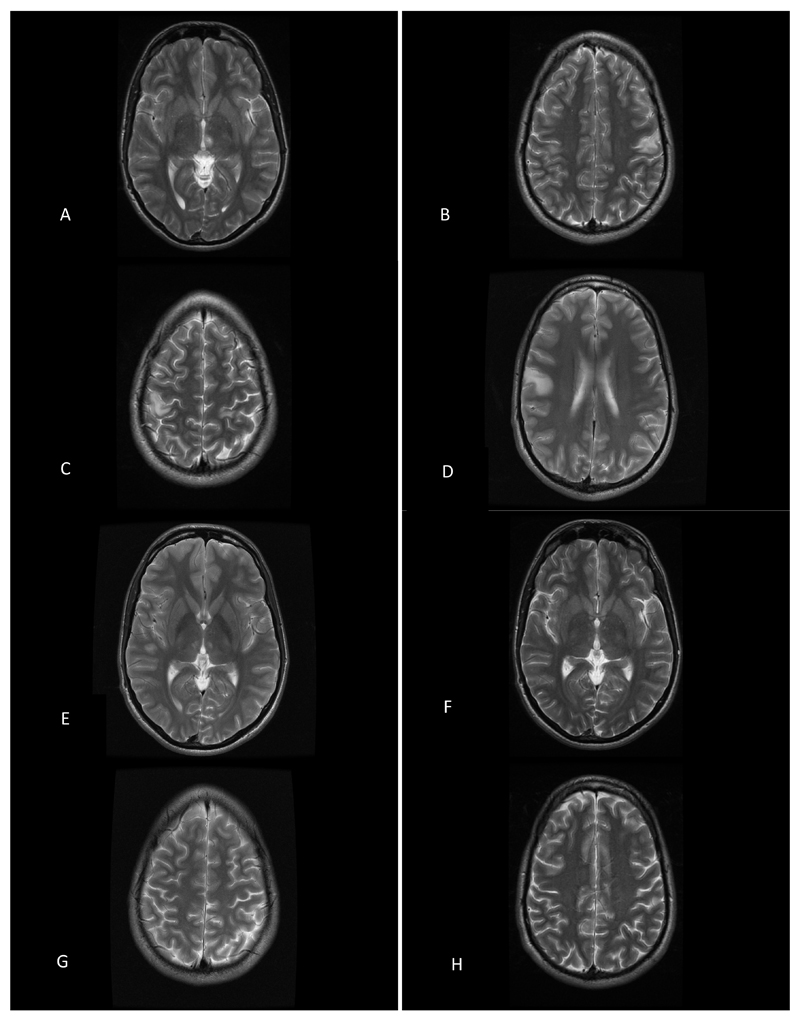
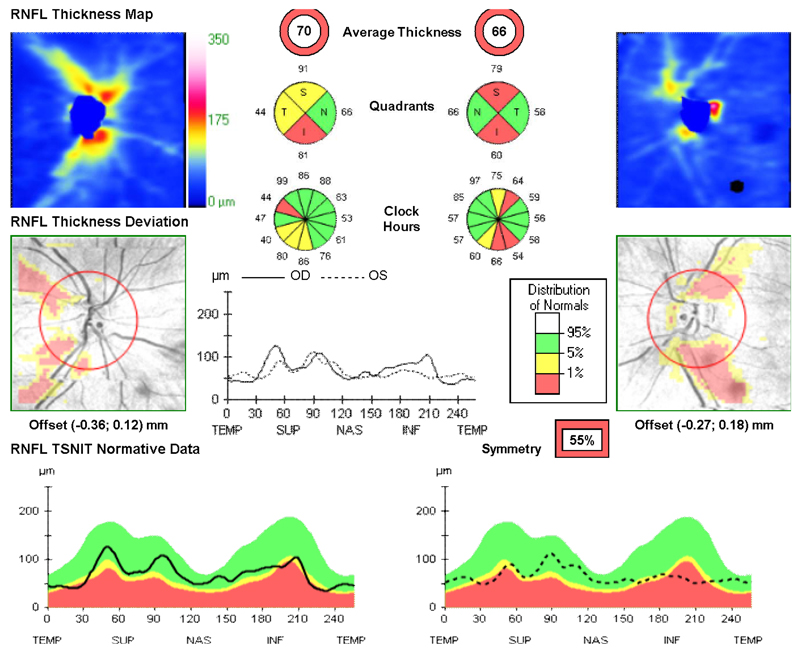
Observations: This case report describes monozygotic male twins with ptosis, optic atrophy, and recent-onset intractable myoclonic epilepsy. The assessment of respiratory chain enzyme activities in the muscle from 1 twin revealed a severe and isolated defect involving mitochondrial complex I. Mitochondrial DNA sequencing revealed a pathogenic m.14487T>C MTND6 mutation, which was present at very high levels of heteroplasmy in muscle (84%) and lower levels in blood (15%), urinary epithelium (75%), and buccal mucosa (58%). Of particular interest, his identical twin was found to harbor very similar levels of the m.14487T>C mutation in his blood, urine, buccal mucosa, and hair follicle DNA samples, while the presence of low levels in the mother's tissues confirmed maternal transmission.
Conclusions and relevance: It was shown that m14487T>C can also cause the unusual combination of optic atrophy, ptosis, and encephalomyopathy leading to intractable seizures. Near-identical heteroplasmy levels in different tissues in both siblings support a nuclear genetic mechanism controlling the tissue segregation of mtDNA mutations.
Figures
Comment in
-
Mitochondrial DNA mutation load: chance or destiny?JAMA Neurol. 2013 Dec;70(12):1484-5. doi: 10.1001/jamaneurol.2013.4401. JAMA Neurol. 2013. PMID: 24126438 Free PMC article. No abstract available.
References
-
- Dermaut B, Seneca S, Dom L, et al. Progressive myoclonic epilepsy as an adult-onset manifestation of Leigh syndrome due to m.14487T>C. J Neurol Neurosurg Psychiatry. 2010;81:90–93. - PubMed
-
- Esteitie N, Hinttala R, Wibom R, et al. Secondary metabolic effects in complex I deficiency. Ann Neurol. 2005;58(4):544–552. - PubMed
-
- Leshinsky-Silver E, Shuvalov R, Inbar S, Cohen S, Lev D, Lerman-Sagie T. Juvenile Leigh syndrome, optic atrophy, ataxia, dystonia, and epilepsy due to T14487C mutation in the mtDNA-ND6 gene: a mitochondrial syndrome presenting from birth to adolescence. J Child Neurol. 2011;26(4):476–481. - PubMed
-
- Malfatti E, Bugiani M, Invernizzi F, et al. Novel mutations of ND genes in complex I deficiency associated with mitochondrial encephalopathy. Brain. 2007;130:1894–1904. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
