

Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States
- PMID: 24129257
- PMCID: PMC3812708
- DOI: 10.1128/mBio.00737-13
Origin, evolution, and genotyping of emergent porcine epidemic diarrhea virus strains in the United States
Abstract
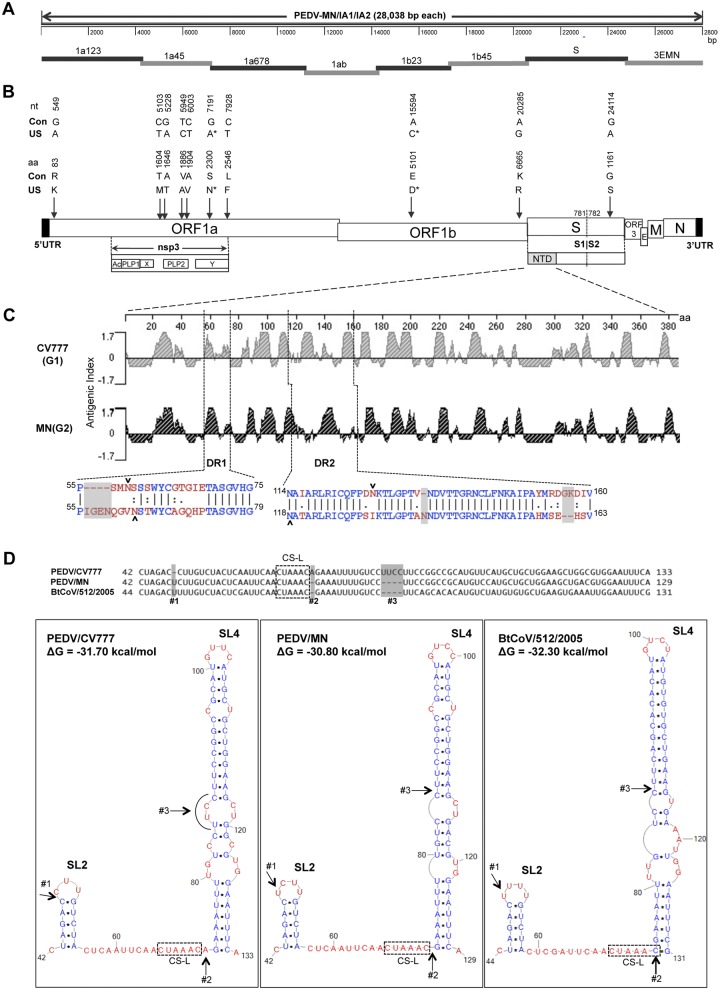
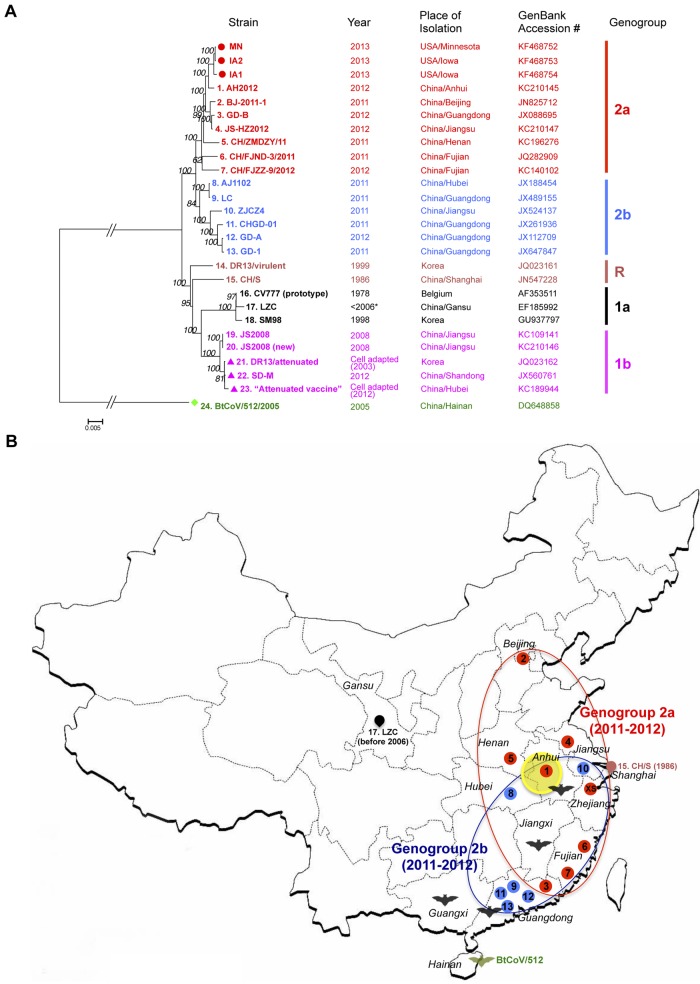
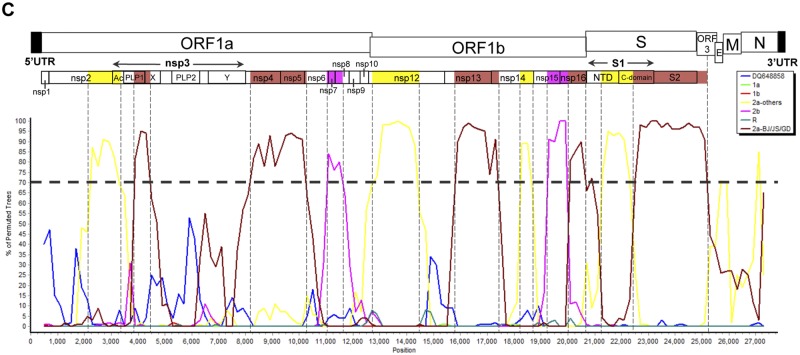
Coronaviruses are known to infect humans and other animals and cause respiratory and gastrointestinal diseases. Here we report the emergence of porcine epidemic diarrhea virus (PEDV) in the United States and determination of its origin, evolution, and genotypes based on temporal and geographical evidence. Histological lesions in small intestine sections of affected pigs and the complete genomic sequences of three emergent strains of PEDV isolated from outbreaks in Minnesota and Iowa were characterized. Genetic and phylogenetic analyses of the three U.S. strains revealed a close relationship with Chinese PEDV strains and their likely Chinese origin. The U.S. PEDV strains underwent evolutionary divergence, which can be classified into two sublineages. The three emergent U.S. strains are most closely related to a strain isolated in 2012 from Anhui Province in China, which might be the result of multiple recombination events between different genetic lineages or sublineages of PEDV. Molecular clock analysis of the divergent time based on the complete genomic sequences is consistent with the actual time difference, approximately 2 to 3 years, of the PED outbreaks between China (December 2010) and the United States (May 2013). The finding that the emergent U.S. PEDV strains share unique genetic features at the 5'-untranslated region with a bat coronavirus provided further support of the evolutionary origin of PEDV from bats and potential cross-species transmission. The data from this study have important implications for understanding the ongoing PEDV outbreaks in the United States and will guide future efforts to develop effective preventive and control measures against PEDV.
Importance: The sudden emergence of porcine epidemic diarrhea virus (PEDV), a coronavirus, for the first time in the United States causes significant economic and public health concerns. Since its recognition in May 2013, PEDV has rapidly spread across the United States, resulting in high mortality in piglets in more than 17 States now. The ongoing outbreaks of Middle East respiratory syndrome coronavirus in humans from countries in or near the Arabian Peninsula and the historical deadly nature of the 2002 outbreaks of severe acute respiratory syndrome coronavirus create further anxiety over the emergence of PEDV in the United States due to the lack of scientific information about the origin and evolution of this emerging coronavirus. Here we report the detailed genetic characterization, origin, and evolution of emergent PEDV strains in the United States. The results provide much needed information to devise effective preventive and control strategies against PEDV in the United States.
Figures
References
-
- de Groot RJ, Baker SC, Baric R, Enjuanes L, Gorbalenya AE, Holmes KV, Perlman S, Poon L, Rottier PJM, Talbot PJ, Woo PCY, Ziebuhr J. 2011. Coronaviridae, p 806–828 In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. (ed), Virus taxonomy: ninth report of the International Committee on Taxonomy of Viruses. Elsevier Academic, London, United Kingdom
-
- Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, Bai R, Teng JL, Tsang CC, Wang M, Zheng BJ, Chan KH, Yuen KY. 2012. Discovery of seven novel mammalian and avian coronaviruses in the genus Deltacoronavirus supports bat coronaviruses as the gene source of Alphacoronavirus and Betacoronavirus and avian coronaviruses as the gene source of Gammacoronavirus and Deltacoronavirus. J. Virol. 86:3995–4008 - PMC - PubMed
-
- van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus AD, Haagmans BL, Gorbalenya AE, Snijder EJ, Fouchier RA. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3(6):e00473-12. 10.1128/mBio.00473-12 - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
