

Correlating structure and function of drug-metabolizing enzymes: progress and ongoing challenges
- PMID: 24130370
- PMCID: PMC3876788
- DOI: 10.1124/dmd.113.054627
Correlating structure and function of drug-metabolizing enzymes: progress and ongoing challenges
Abstract
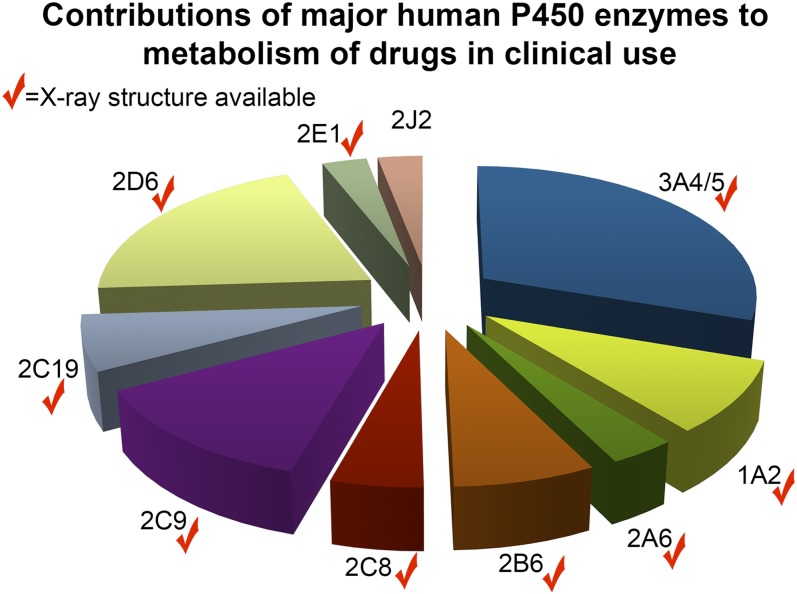
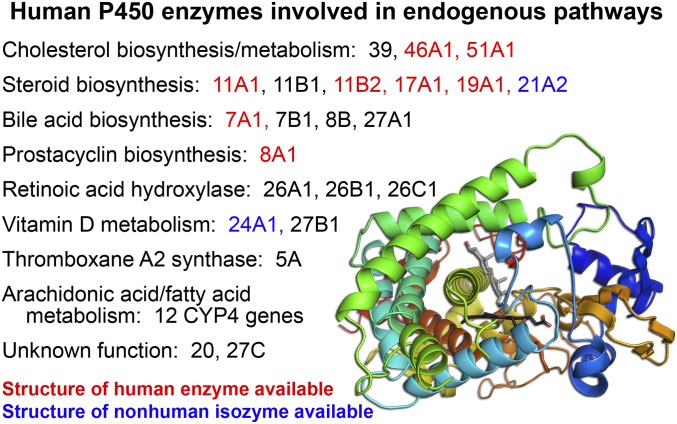
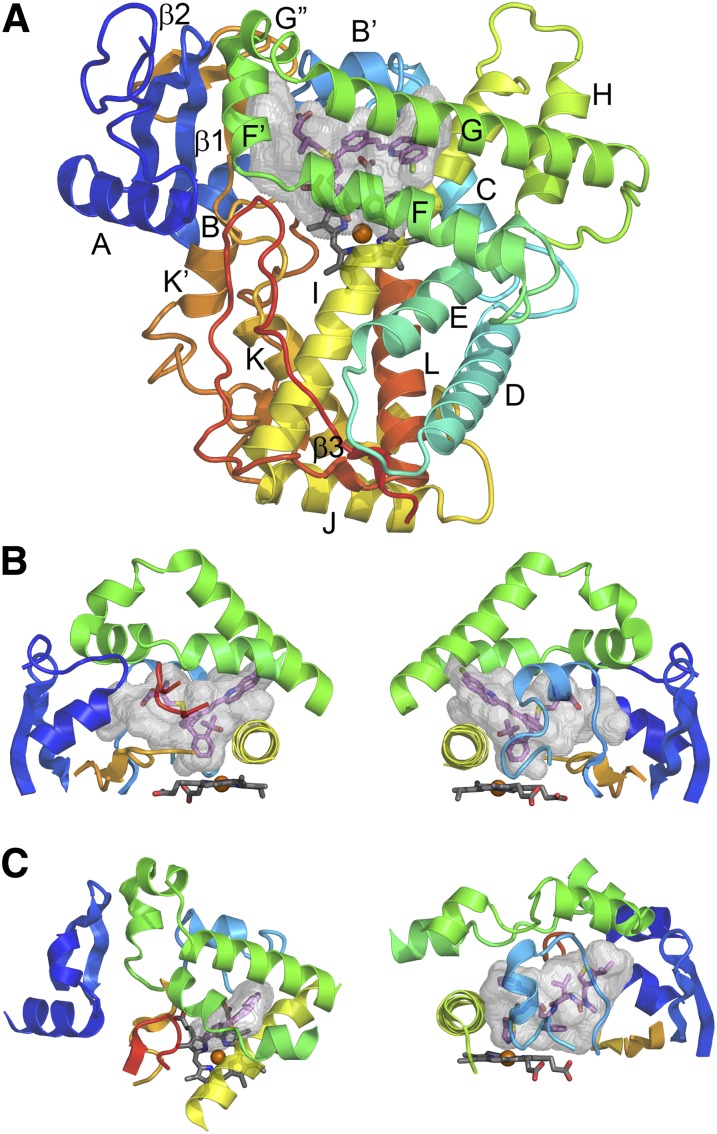
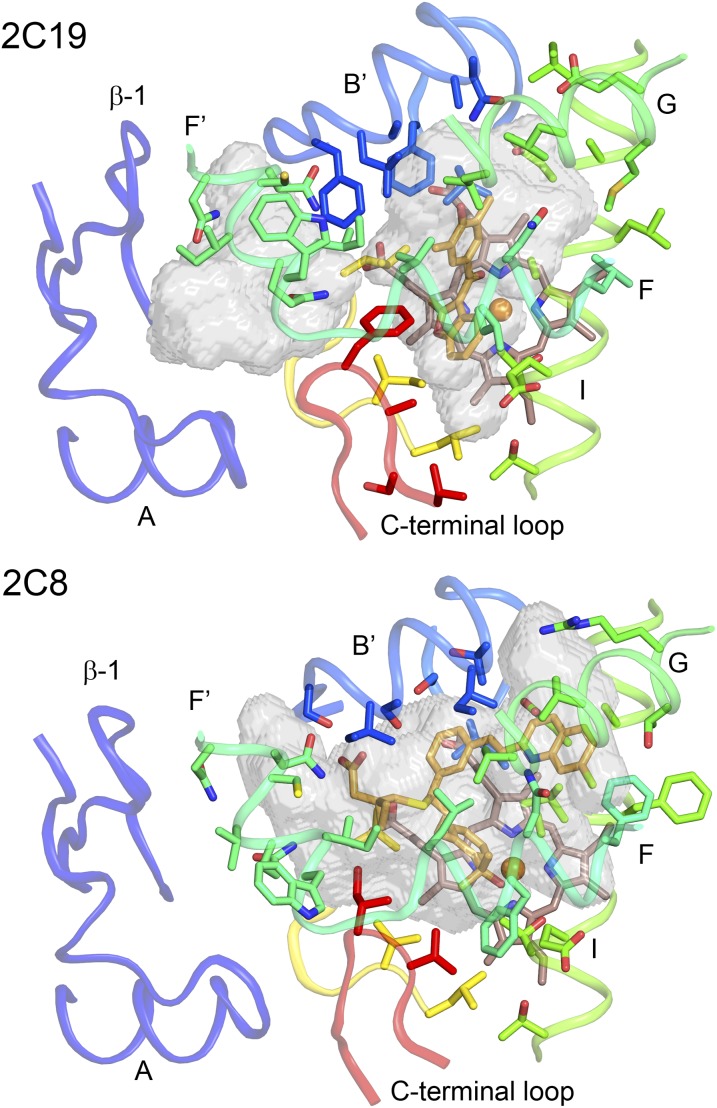
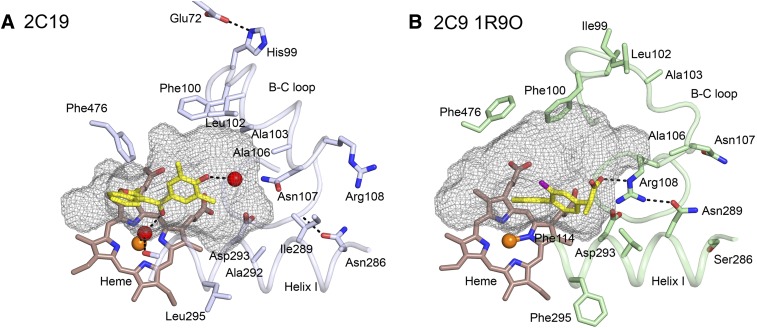
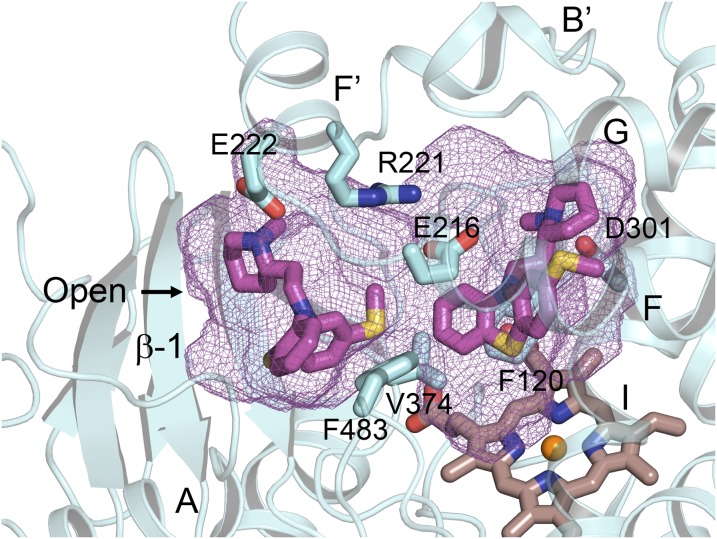
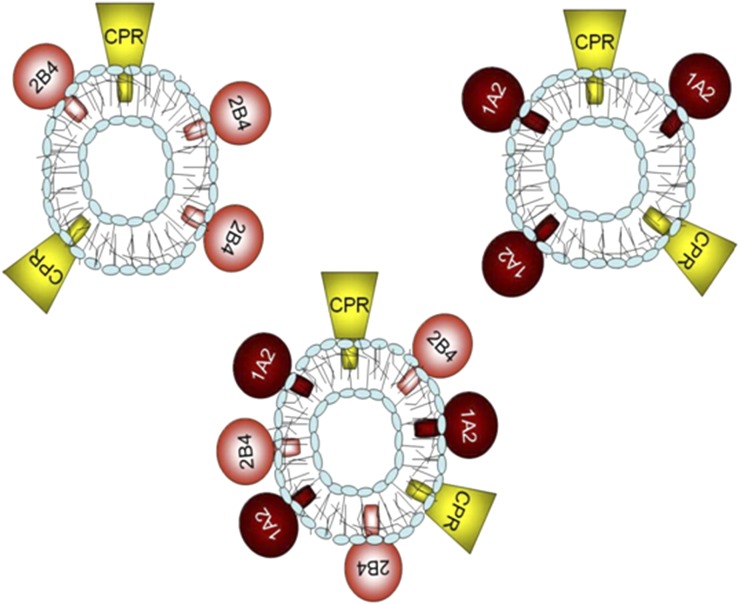
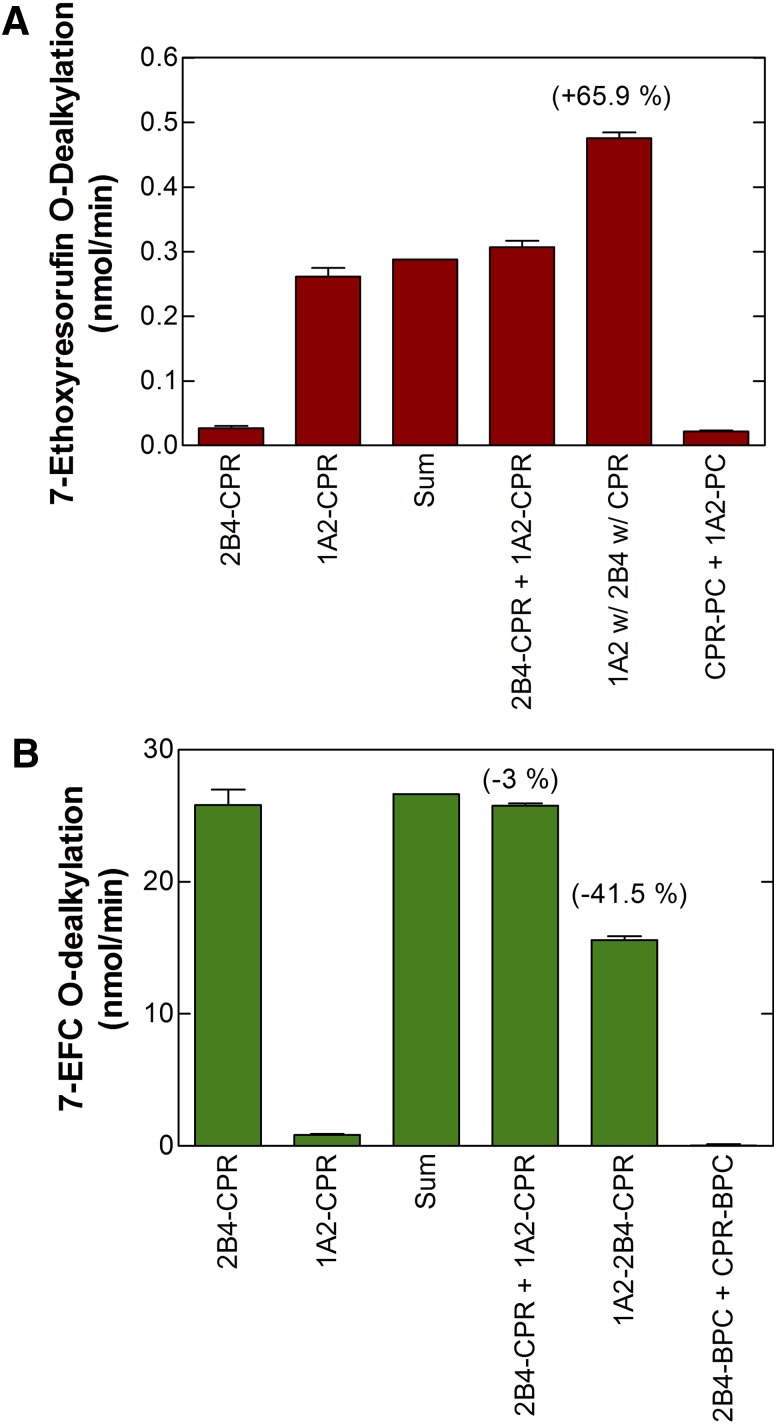
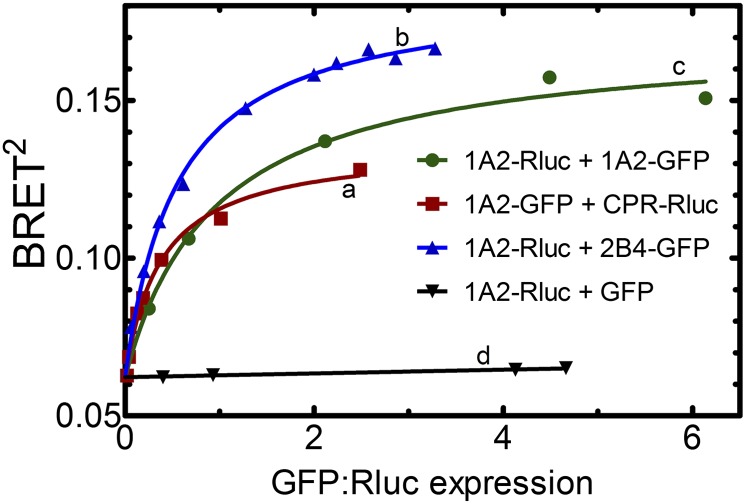
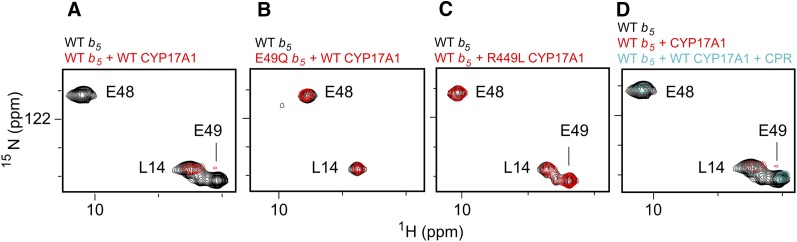
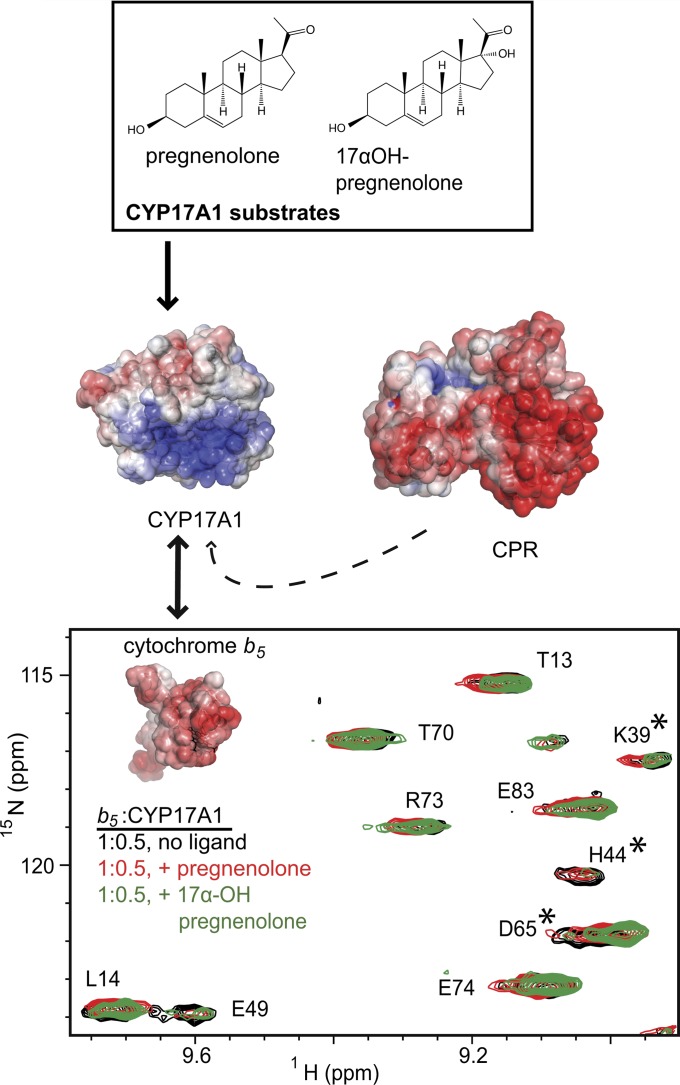
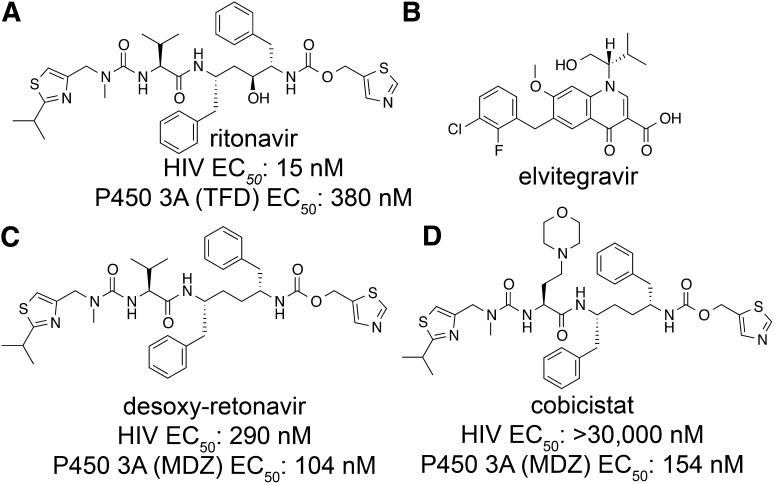
This report summarizes a symposium sponsored by the American Society for Pharmacology and Experimental Therapeutics at Experimental Biology held April 20-24 in Boston, MA. Presentations discussed the status of cytochrome P450 (P450) knowledge, emphasizing advances and challenges in relating structure with function and in applying this information to drug design. First, at least one structure of most major human drug-metabolizing P450 enzymes is known. However, the flexibility of these active sites can limit the predictive value of one structure for other ligands. A second limitation is our coarse-grain understanding of P450 interactions with membranes, other P450 enzymes, NADPH-cytochrome P450 reductase, and cytochrome b5. Recent work has examined differential P450 interactions with reductase in mixed P450 systems and P450:P450 complexes in reconstituted systems and cells, suggesting another level of functional control. In addition, protein nuclear magnetic resonance is a new approach to probe these protein/protein interactions, identifying interacting b5 and P450 surfaces, showing that b5 and reductase binding are mutually exclusive, and demonstrating ligand modulation of CYP17A1/b5 interactions. One desired outcome is the application of such information to control drug metabolism and/or design selective P450 inhibitors. A final presentation highlighted development of a CYP3A4 inhibitor that slows clearance of human immunodeficiency virus drugs otherwise rapidly metabolized by CYP3A4. Although understanding P450 structure/function relationships is an ongoing challenge, translational advances will benefit from continued integration of existing and new biophysical approaches.
Figures
References
-
- Akhtar MK, Kelly SL, Kaderbhai MA. (2005) Cytochrome b(5) modulation of 17alpha hydroxylase and 17-20 lyase (CYP17) activities in steroidogenesis. J Endocrinol 187:267–274 - PubMed
-
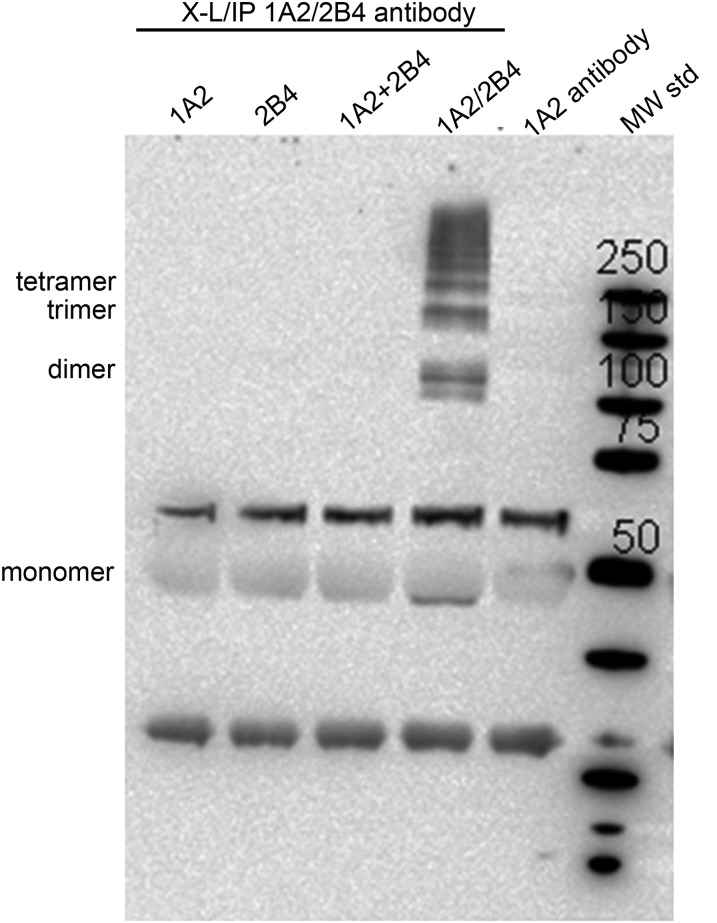
- Backes WL, Batie CJ, Cawley GF. (1998) Interactions among P450 enzymes when combined in reconstituted systems: formation of a 2B4-1A2 complex with a high affinity for NADPH-cytochrome P450 reductase. Biochemistry 37:12852–12859 - PubMed
-
- Bass MB, Paulsen MD, Ornstein RL. (1992) Substrate mobility in a deeply buried active site: analysis of norcamphor bound to cytochrome P-450cam as determined by a 201-psec molecular dynamics simulation. Proteins 13:26–37 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
