

Feature selection methods for identifying genetic determinants of host species in RNA viruses
- PMID: 24130470
- PMCID: PMC3794897
- DOI: 10.1371/journal.pcbi.1003254
Feature selection methods for identifying genetic determinants of host species in RNA viruses
Abstract
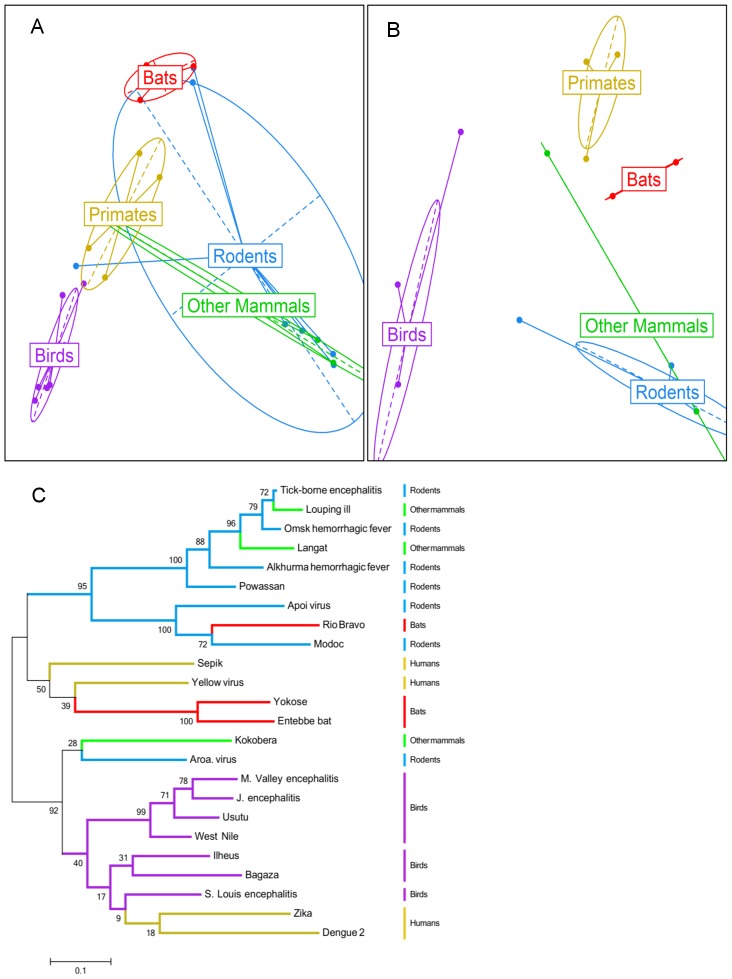
Despite environmental, social and ecological dependencies, emergence of zoonotic viruses in human populations is clearly also affected by genetic factors which determine cross-species transmission potential. RNA viruses pose an interesting case study given their mutation rates are orders of magnitude higher than any other pathogen--as reflected by the recent emergence of SARS and Influenza for example. Here, we show how feature selection techniques can be used to reliably classify viral sequences by host species, and to identify the crucial minority of host-specific sites in pathogen genomic data. The variability in alleles at those sites can be translated into prediction probabilities that a particular pathogen isolate is adapted to a given host. We illustrate the power of these methods by: 1) identifying the sites explaining SARS coronavirus differences between human, bat and palm civet samples; 2) showing how cross species jumps of rabies virus among bat populations can be readily identified; and 3) de novo identification of likely functional influenza host discriminant markers.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Accelerated viral dynamics in bat cell lines, with implications for zoonotic emergence.Elife. 2020 Feb 3;9:e48401. doi: 10.7554/eLife.48401. Elife. 2020. PMID: 32011232 Free PMC article.
-
Bats as Viral Reservoirs.Annu Rev Virol. 2016 Sep 29;3(1):77-99. doi: 10.1146/annurev-virology-110615-042203. Epub 2016 Aug 22. Annu Rev Virol. 2016. PMID: 27578437 Review.
-
Rates of viral evolution are linked to host geography in bat rabies.PLoS Pathog. 2012;8(5):e1002720. doi: 10.1371/journal.ppat.1002720. Epub 2012 May 17. PLoS Pathog. 2012. PMID: 22615575 Free PMC article.
-
The Species-Specific 282 Residue in the PB2 Subunit of the Polymerase Regulates RNA Synthesis and Replication of Influenza A Viruses Infecting Bat and Nonbat Hosts.J Virol. 2022 Mar 9;96(5):e0219021. doi: 10.1128/jvi.02190-21. Epub 2022 Jan 19. J Virol. 2022. PMID: 35044213 Free PMC article.
-
[Bats and Viruses: complex relationships].Bull Soc Pathol Exot. 2015 Oct;108(4):272-89. doi: 10.1007/s13149-015-0448-z. Epub 2015 Sep 1. Bull Soc Pathol Exot. 2015. PMID: 26330152 Free PMC article. Review.
Cited by
-
Fermented Seeds ("Zgougou") from Aleppo Pine as a Novel Source of Potentially Probiotic Lactic Acid Bacteria.Microorganisms. 2019 Dec 17;7(12):709. doi: 10.3390/microorganisms7120709. Microorganisms. 2019. PMID: 31861080 Free PMC article.
-
Existing Host Range Mutations Constrain Further Emergence of RNA Viruses.J Virol. 2019 Feb 5;93(4):e01385-18. doi: 10.1128/JVI.01385-18. Print 2019 Feb 15. J Virol. 2019. PMID: 30463962 Free PMC article.
-
Lineage structure of Streptococcus pneumoniae may be driven by immune selection on the groEL heat-shock protein.Sci Rep. 2017 Aug 22;7(1):9023. doi: 10.1038/s41598-017-08990-z. Sci Rep. 2017. PMID: 28831154 Free PMC article.
-
Comparative studies of alignment, alignment-free and SVM based approaches for predicting the hosts of viruses based on viral sequences.Sci Rep. 2018 Jul 3;8(1):10032. doi: 10.1038/s41598-018-28308-x. Sci Rep. 2018. PMID: 29968780 Free PMC article.
-
K-Pax2: Bayesian identification of cluster-defining amino acid positions in large sequence datasets.Microb Genom. 2015 Jul 15;1(1):e000025. doi: 10.1099/mgen.0.000025. eCollection 2015 Jul. Microb Genom. 2015. PMID: 28348810 Free PMC article.
References
-
- Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, et al. (2000) Nipah virus: a recently emergent deadly paramyxovirus. Science 288: 1432–1435. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
