

Genome-wide high resolution parental-specific DNA and histone methylation maps uncover patterns of imprinting regulation in maize
- PMID: 24131563
- PMCID: PMC3875858
- DOI: 10.1101/gr.155879.113
Genome-wide high resolution parental-specific DNA and histone methylation maps uncover patterns of imprinting regulation in maize
Abstract
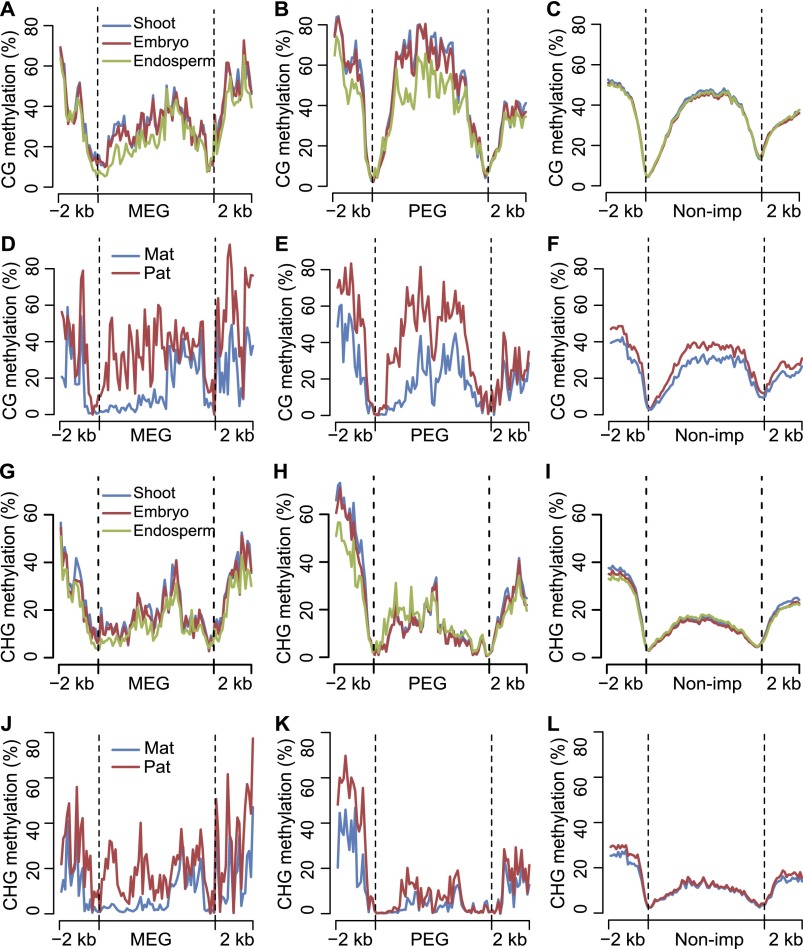
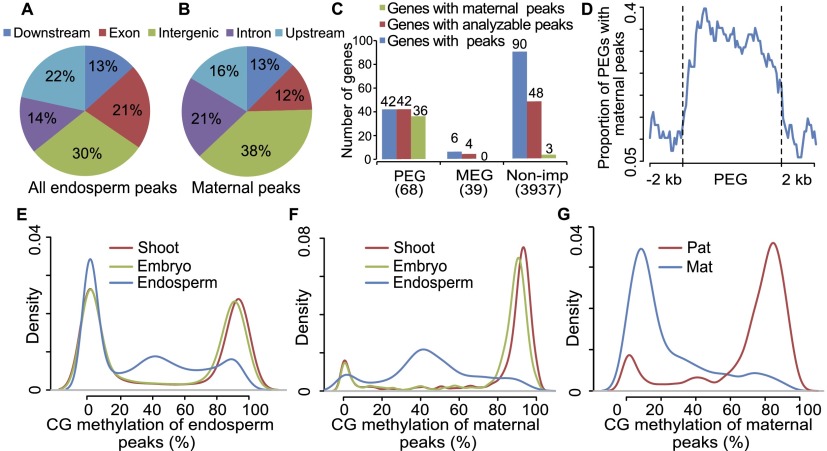
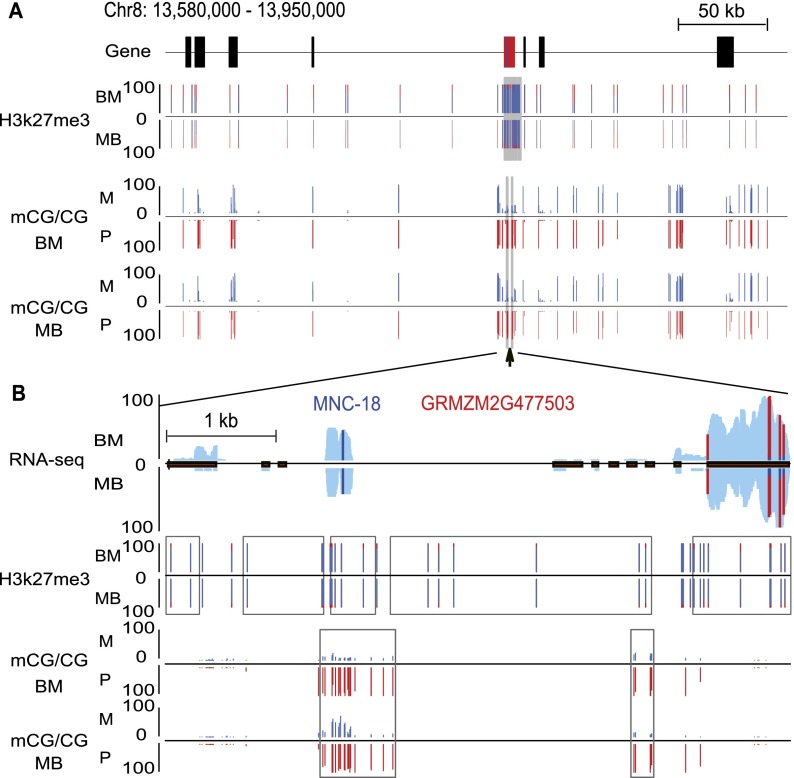
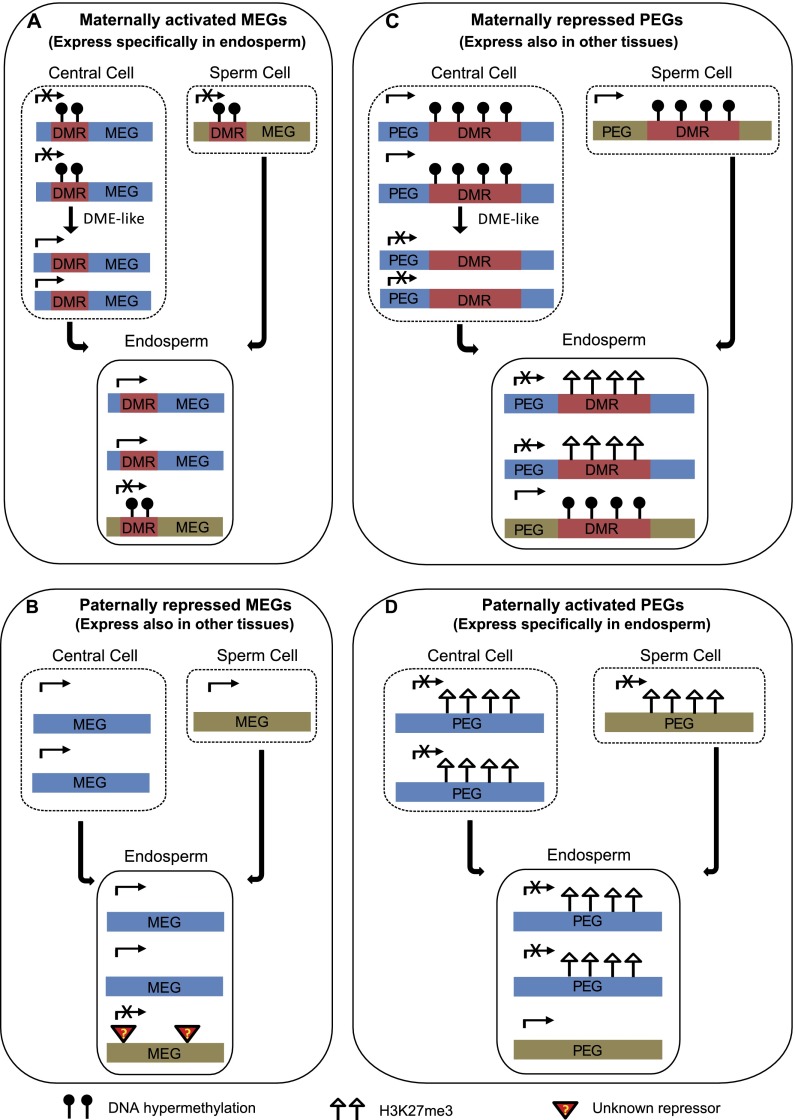
Genetic imprinting is a specific epigenetic phenomenon in which a subset of genes is expressed depending on their parent-of-origin. Two types of chromatin modifications, DNA methylation and histone modification, are generally believed to be involved in the regulation of imprinting. However, the genome-wide correlation between allele-specific chromatin modifications and imprinted gene expression in maize remains elusive. Here we report genome-wide high resolution allele-specific maps of DNA methylation and histone H3 lysine 27 trimethylation (H3K27me3) in maize endosperm. For DNA methylation, thousands of parent-of-origin dependent differentially methylated regions (pDMRs) were identified. All pDMRs were uniformly paternally hypermethylated and maternally hypomethylated. We also identified 1131 allele-specific H3K27me3 peaks that are preferentially present in the maternal alleles. Maternally expressed imprinted genes (MEGs) and paternally expressed imprinted genes (PEGs) had different patterns of allele-specific DNA methylation and H3K27me3. Allele-specific expression of MEGs was not directly related to allele-specific H3K27me3, and only a subset of MEGs was associated with maternal-specific DNA demethylation, which was primarily located in the upstream and 5' portion of gene body regions. In contrast, allele-specific expression of a majority of PEGs was related to maternal-specific H3K27me3, with a subgroup of PEGs also associated with maternal-specific DNA demethylation. Both pDMRs and maternal H3K27me3 peaks associated with PEGs are enriched in gene body regions. Our results indicate highly complex patterns of regulation on genetic imprinting in maize endosperm.
Figures
References
-
- Barlow DP 2011. Genomic imprinting: A mammalian epigenetic discovery model. Annu Rev Genet 45: 379–403 - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases