

Mass spectrometry coupled experiments and protein structure modeling methods
- PMID: 24132151
- PMCID: PMC3821635
- DOI: 10.3390/ijms141020635
Mass spectrometry coupled experiments and protein structure modeling methods
Abstract
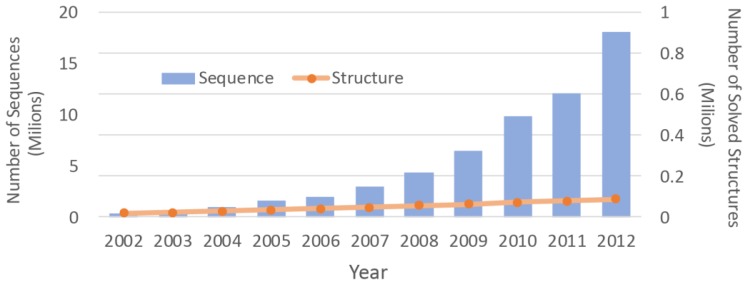
With the accumulation of next generation sequencing data, there is increasing interest in the study of intra-species difference in molecular biology, especially in relation to disease analysis. Furthermore, the dynamics of the protein is being identified as a critical factor in its function. Although accuracy of protein structure prediction methods is high, provided there are structural templates, most methods are still insensitive to amino-acid differences at critical points that may change the overall structure. Also, predicted structures are inherently static and do not provide information about structural change over time. It is challenging to address the sensitivity and the dynamics by computational structure predictions alone. However, with the fast development of diverse mass spectrometry coupled experiments, low-resolution but fast and sensitive structural information can be obtained. This information can then be integrated into the structure prediction process to further improve the sensitivity and address the dynamics of the protein structures. For this purpose, this article focuses on reviewing two aspects: the types of mass spectrometry coupled experiments and structural data that are obtainable through those experiments; and the structure prediction methods that can utilize these data as constraints. Also, short review of current efforts in integrating experimental data in the structural modeling is provided.
Figures
References
-
- Apweiler R., Bairoch A., Wu C.H. Protein sequence databases. Curr. Opin. Chem. Biol. 2004;8:76–80. - PubMed
-
- Gao X. Ph.D. Thesis. University of Waterloo; Waterloo, ON, Canada: 2009. Towards Automating Protein Structure Determination from NMR Data.
-
- Skolnick J., Zhang Y., Arakaki A.K., Kolinski A., Boniecki M., Szilágyi A., Kihara D. TOUCHSTONE: A unified approach to protein structure prediction. Proteins. 2003;53:469–479. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
