

Mutational landscape and significance across 12 major cancer types
- PMID: 24132290
- PMCID: PMC3927368
- DOI: 10.1038/nature12634
Mutational landscape and significance across 12 major cancer types
Abstract
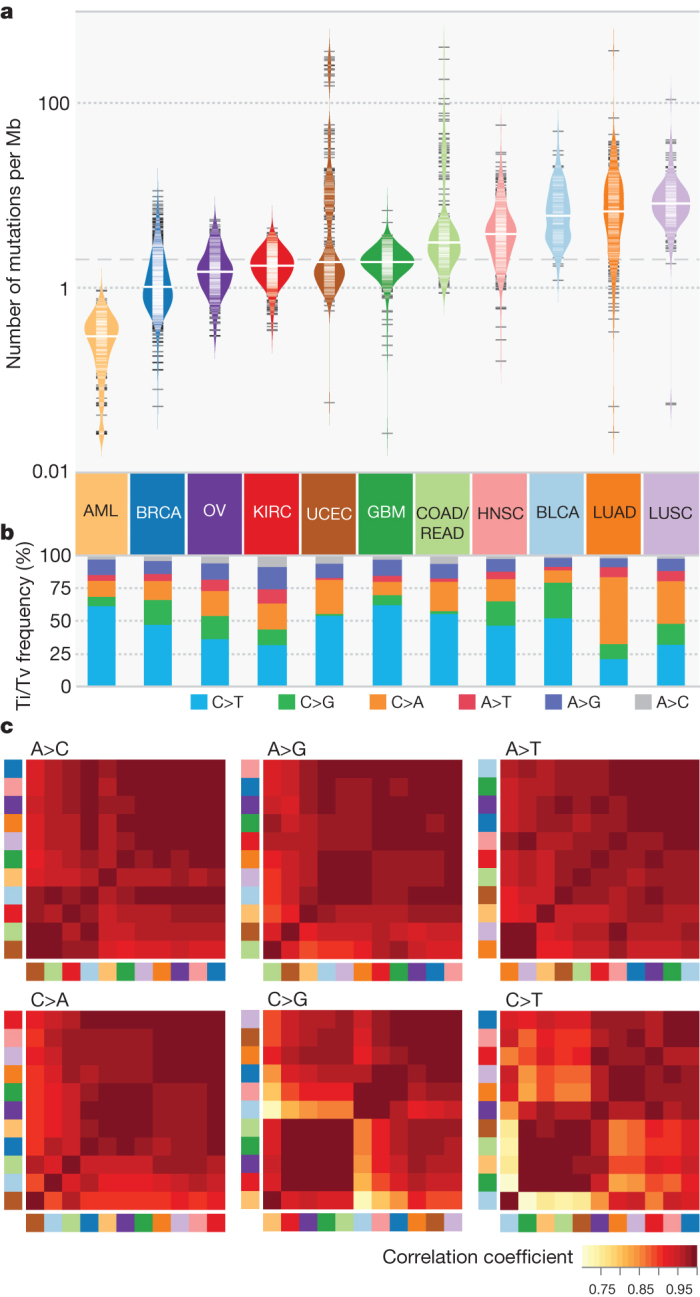
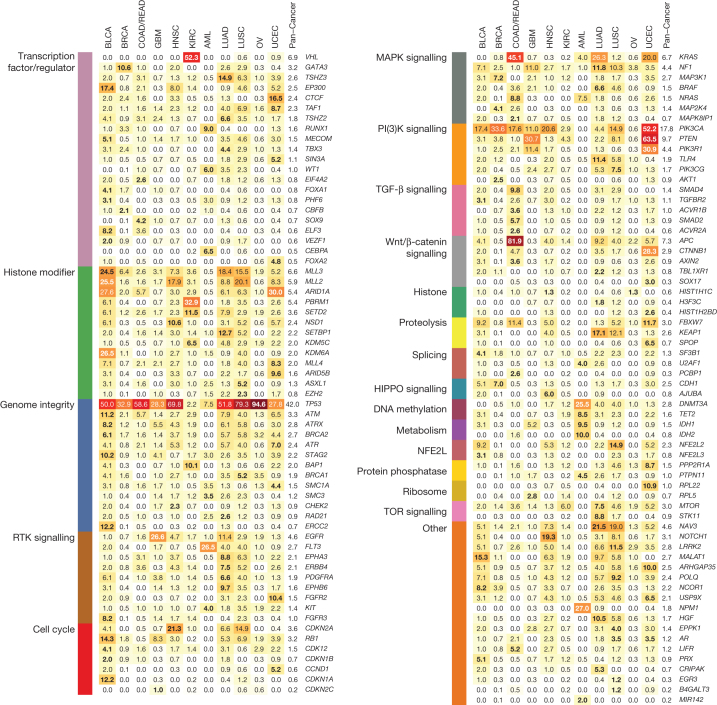
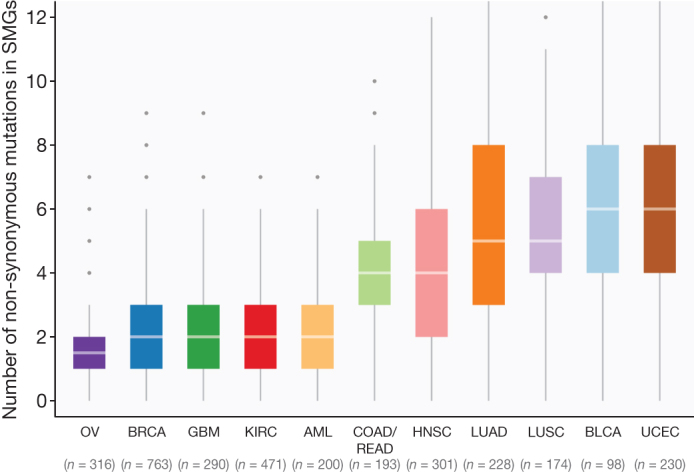
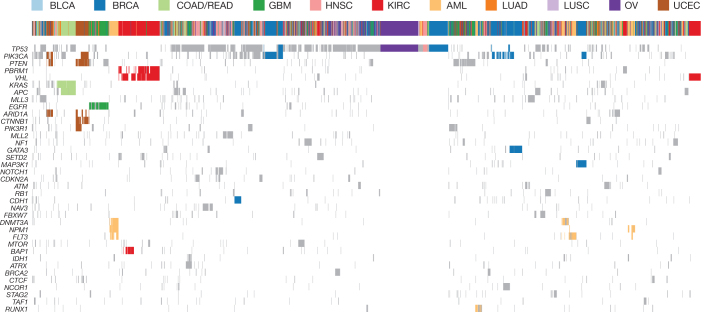
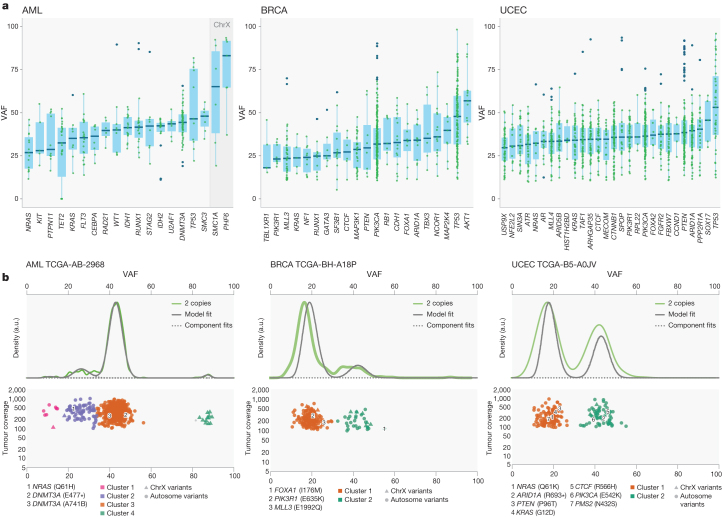
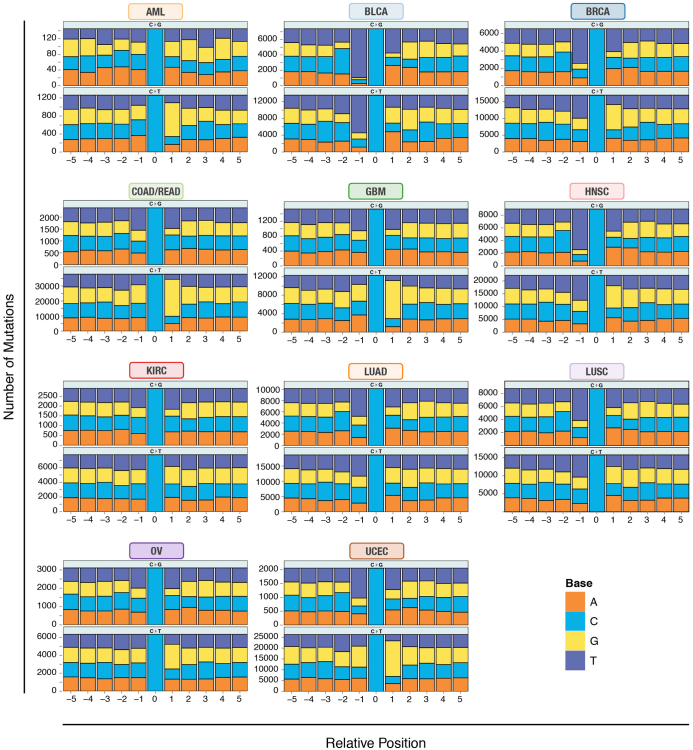
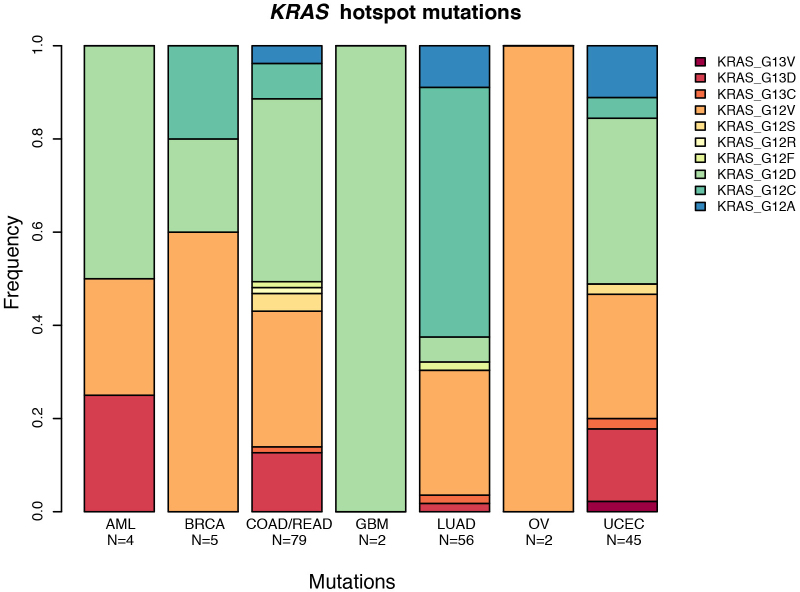
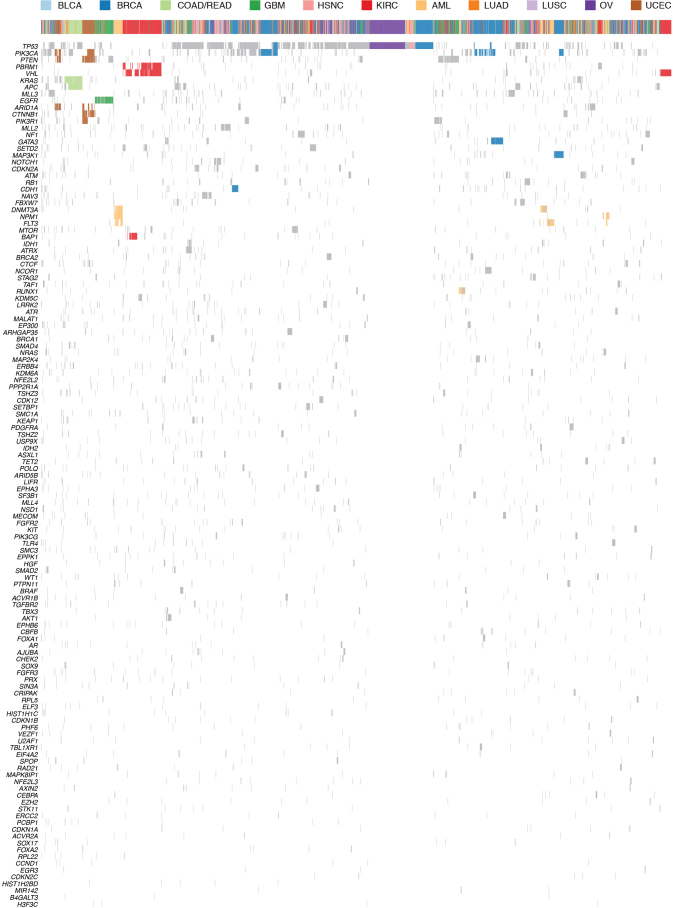
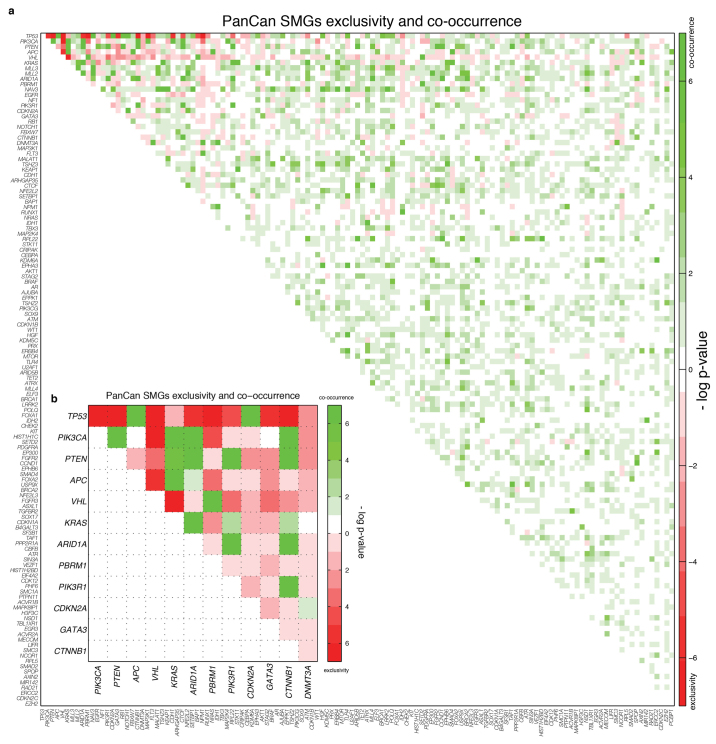
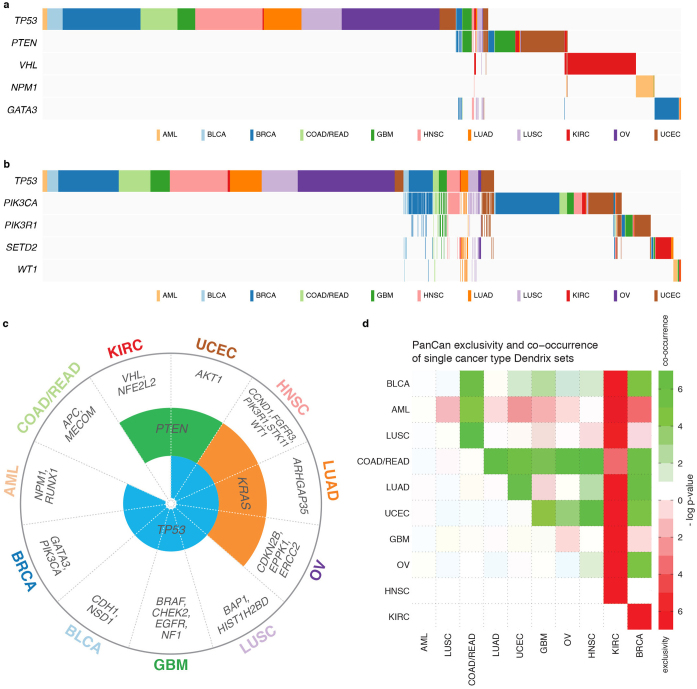
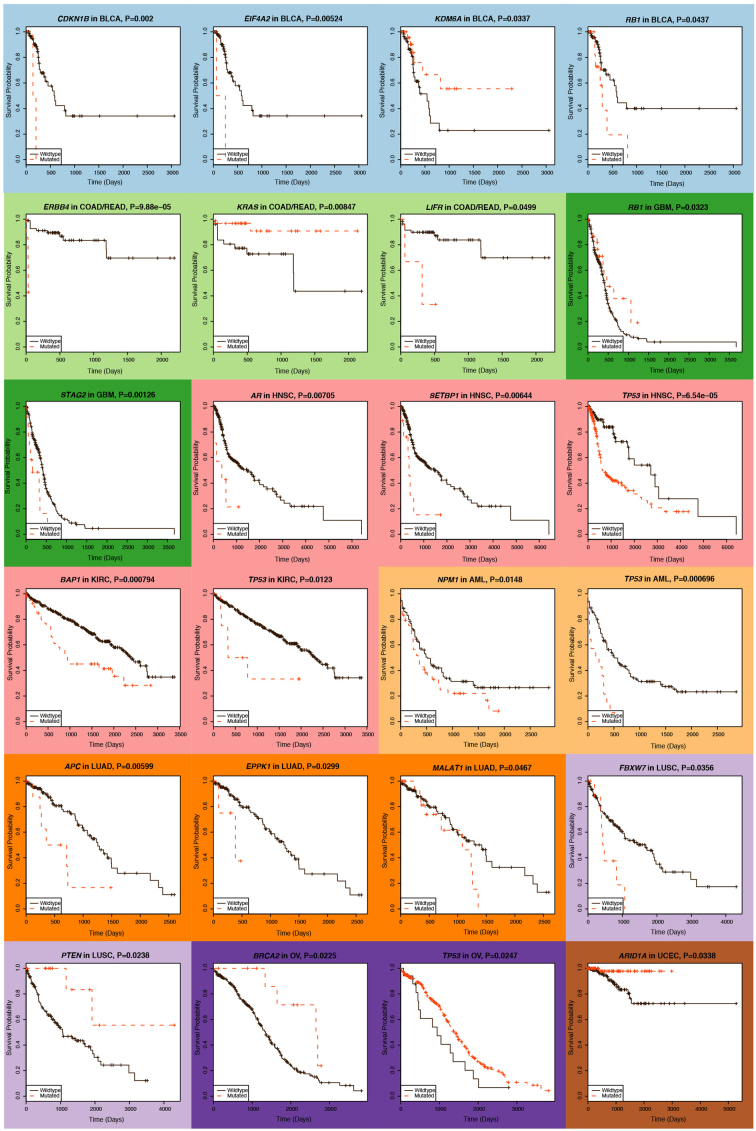
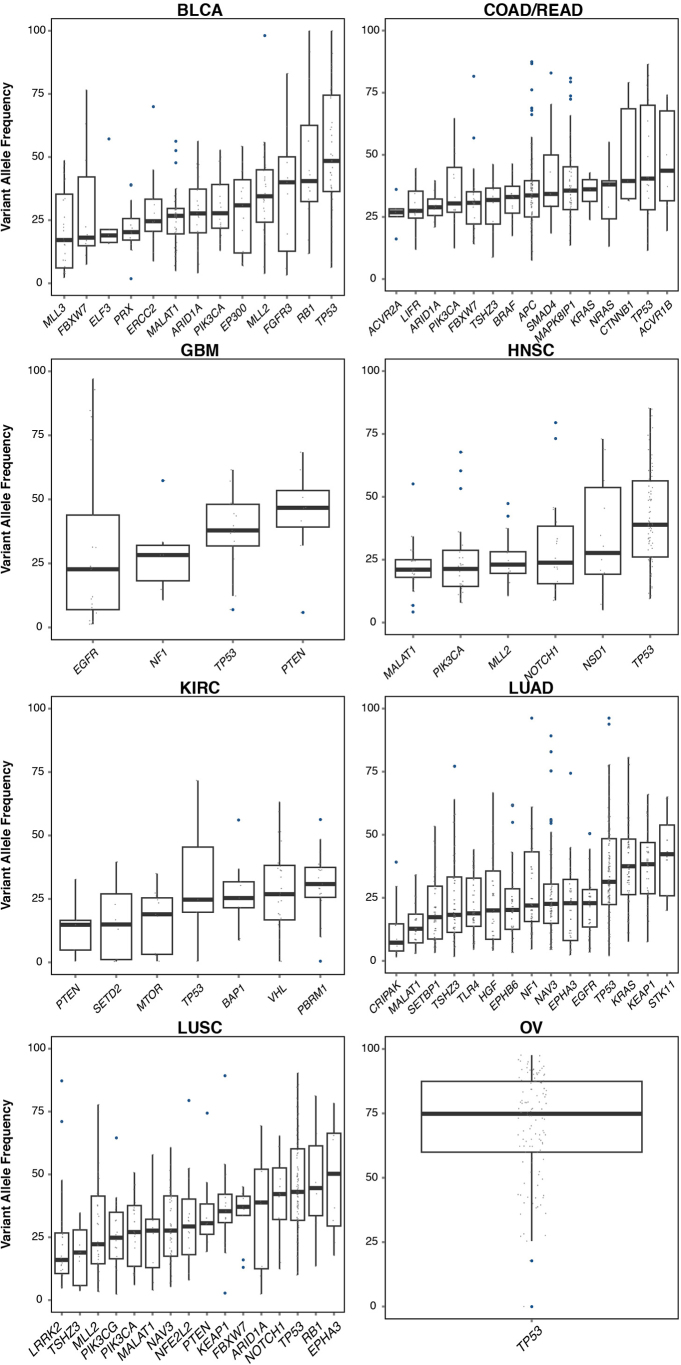
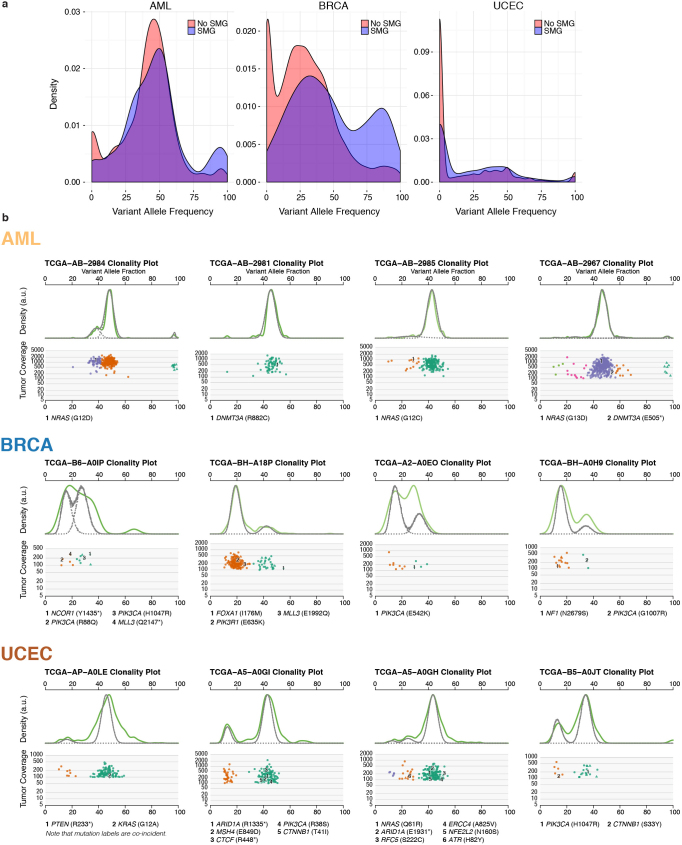
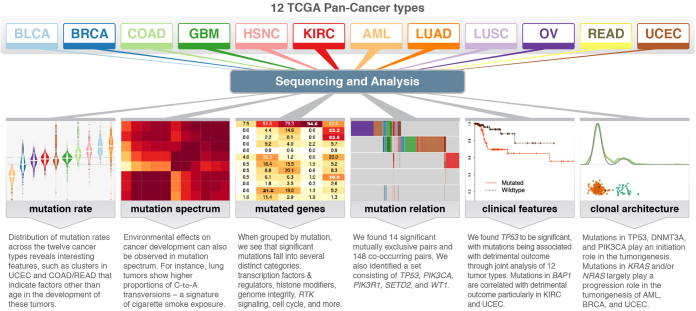
The Cancer Genome Atlas (TCGA) has used the latest sequencing and analysis methods to identify somatic variants across thousands of tumours. Here we present data and analytical results for point mutations and small insertions/deletions from 3,281 tumours across 12 tumour types as part of the TCGA Pan-Cancer effort. We illustrate the distributions of mutation frequencies, types and contexts across tumour types, and establish their links to tissues of origin, environmental/carcinogen influences, and DNA repair defects. Using the integrated data sets, we identified 127 significantly mutated genes from well-known (for example, mitogen-activated protein kinase, phosphatidylinositol-3-OH kinase, Wnt/β-catenin and receptor tyrosine kinase signalling pathways, and cell cycle control) and emerging (for example, histone, histone modification, splicing, metabolism and proteolysis) cellular processes in cancer. The average number of mutations in these significantly mutated genes varies across tumour types; most tumours have two to six, indicating that the number of driver mutations required during oncogenesis is relatively small. Mutations in transcriptional factors/regulators show tissue specificity, whereas histone modifiers are often mutated across several cancer types. Clinical association analysis identifies genes having a significant effect on survival, and investigations of mutations with respect to clonal/subclonal architecture delineate their temporal orders during tumorigenesis. Taken together, these results lay the groundwork for developing new diagnostics and individualizing cancer treatment.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Genomics: Comparisons across cancers.Nature. 2013 Oct 17;502(7471):306-7. doi: 10.1038/502306a. Nature. 2013. PMID: 24132284 No abstract available.
-
Cancer genomics: A panoramic view of cancer.Nat Rev Genet. 2013 Nov;14(11):750. doi: 10.1038/nrg3602. Nat Rev Genet. 2013. PMID: 24136505 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
