

Population pharmacokinetic-pharmacodynamic modelling in oncology: a tool for predicting clinical response
- PMID: 24134068
- PMCID: PMC4294077
- DOI: 10.1111/bcp.12258
Population pharmacokinetic-pharmacodynamic modelling in oncology: a tool for predicting clinical response
Abstract
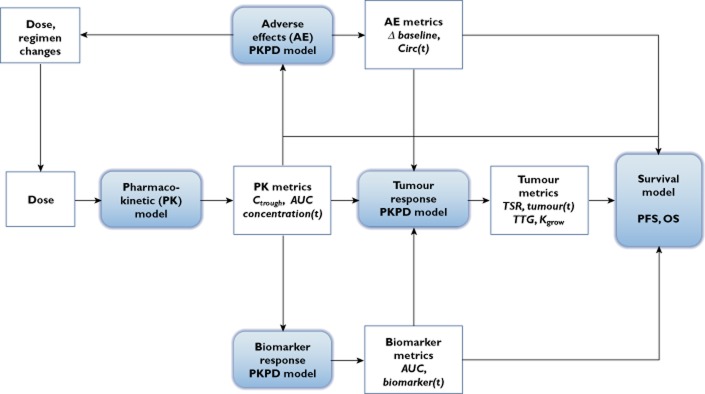
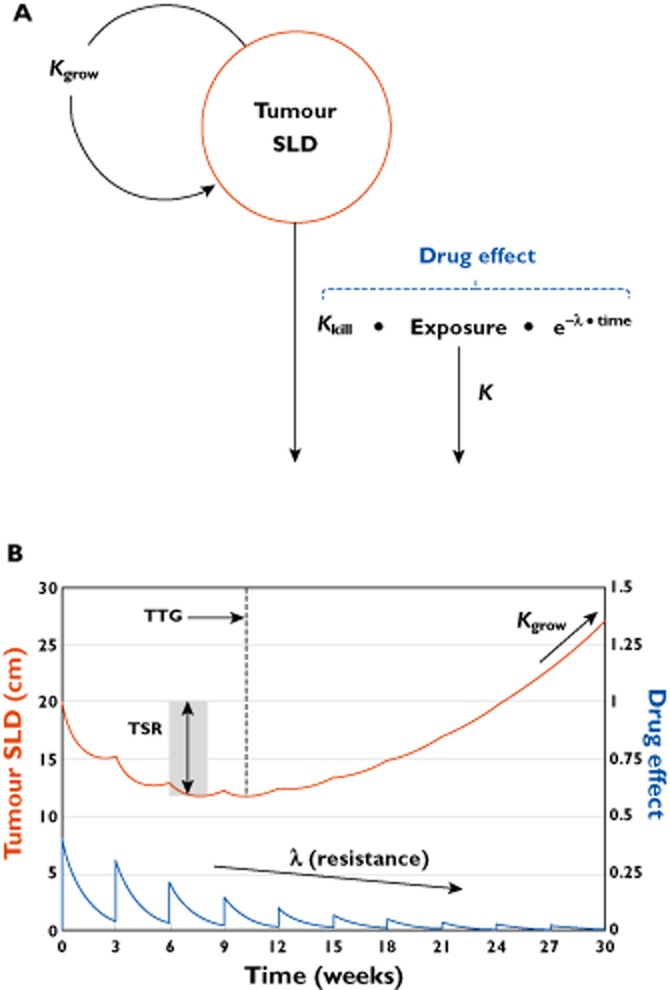
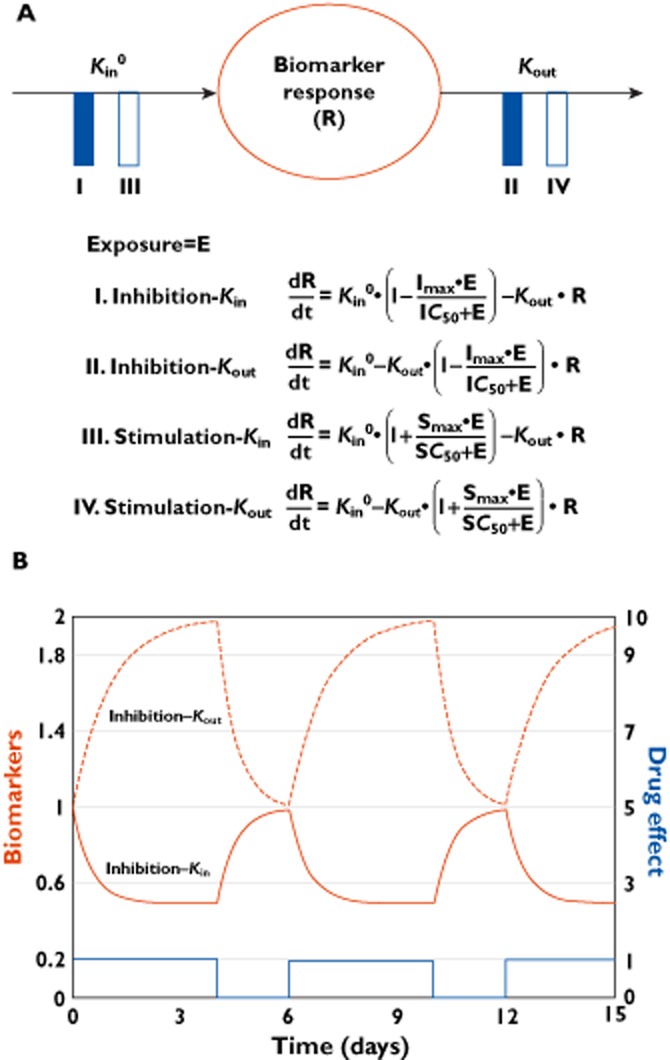
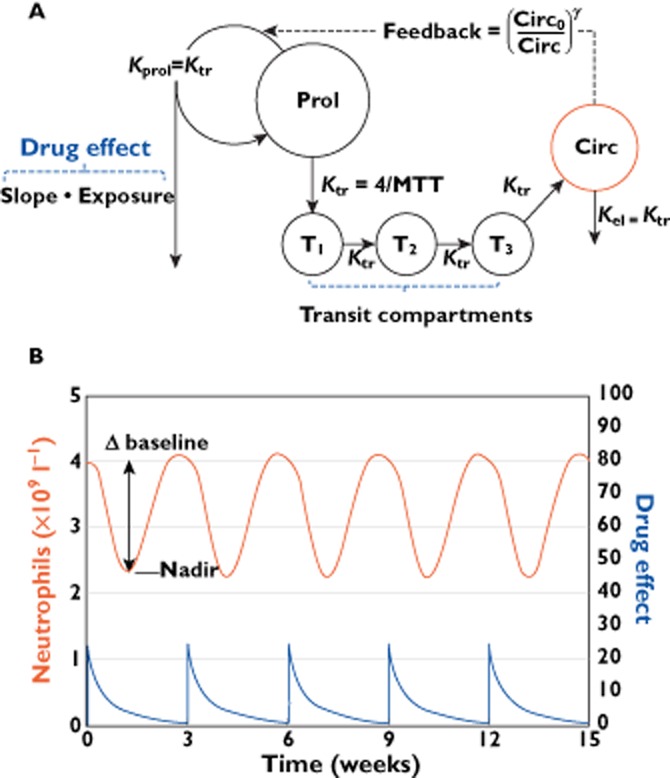
In oncology trials, overall survival (OS) is considered the most reliable and preferred endpoint to evaluate the benefit of drug treatment. Other relevant variables are also collected from patients for a given drug and its indication, and it is important to characterize the dynamic effects and links between these variables in order to improve the speed and efficiency of clinical oncology drug development. However, the drug-induced effects and causal relationships are often difficult to interpret because of temporal differences. To address this, population pharmacokinetic-pharmacodynamic (PKPD) modelling and parametric time-to-event (TTE) models are becoming more frequently applied. Population PKPD and TTE models allow for exploration towards describing the data, understanding the disease and drug action over time, investigating relevance of biomarkers, quantifying patient variability and in designing successful trials. In addition, development of models characterizing both desired and adverse effects in a modelling framework support exploration of risk-benefit of different dosing schedules. In this review, we have summarized population PKPD modelling analyses describing tumour, tumour marker and biomarker responses, as well as adverse effects, from anticancer drug treatment data. Various model-based metrics used to drive PD response and predict OS for oncology drugs and their indications are also discussed.
Keywords: PKPD; biomarkers; oncology; population modelling; time-to-event; tumour.
© 2013 The Authors. British Journal of Clinical Pharmacology published by John Wiley & Sons Ltd on behalf of The British Pharmacological Society.
Figures
References
-
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. - PubMed
-
- FDA. 2007. Guidance for industry. Clinical trial endpoints for the approval of cancer drugs and biologics. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformati... (last accessed 7 November 2013)
-
- Booth CM, Eisenhauer EA. Progression-free survival: meaningful or simply measurable? J Clin Oncol. 2012;30:1030–1033. - PubMed
-
- Bruno R, Claret L. On the use of change in tumor size to predict survival in clinical oncology studies: toward a new paradigm to design and evaluate phase II studies. Clin Pharmacol Ther. 2009;86:136–138. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
