

Developmental and tumoral vascularization is regulated by G protein-coupled receptor kinase 2
- PMID: 24135140
- PMCID: PMC3809778
- DOI: 10.1172/JCI67333
Developmental and tumoral vascularization is regulated by G protein-coupled receptor kinase 2
Abstract
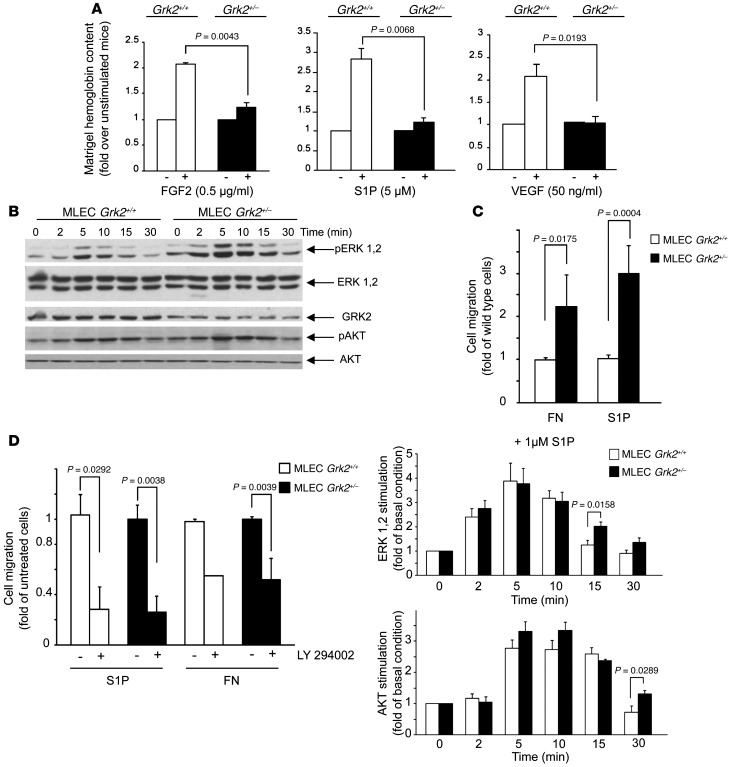
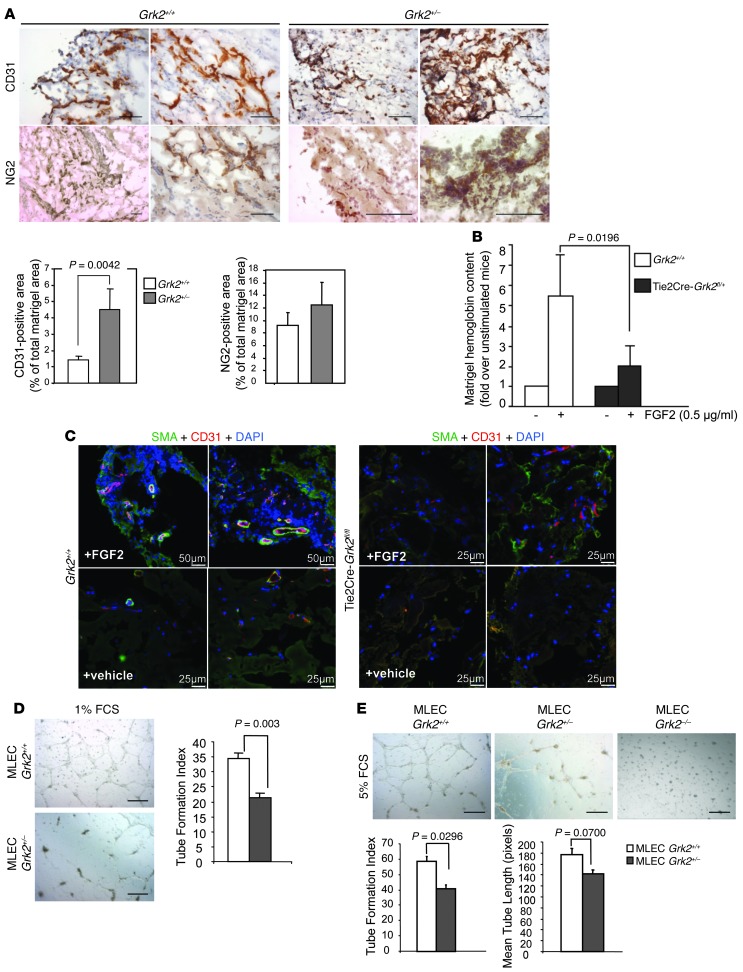
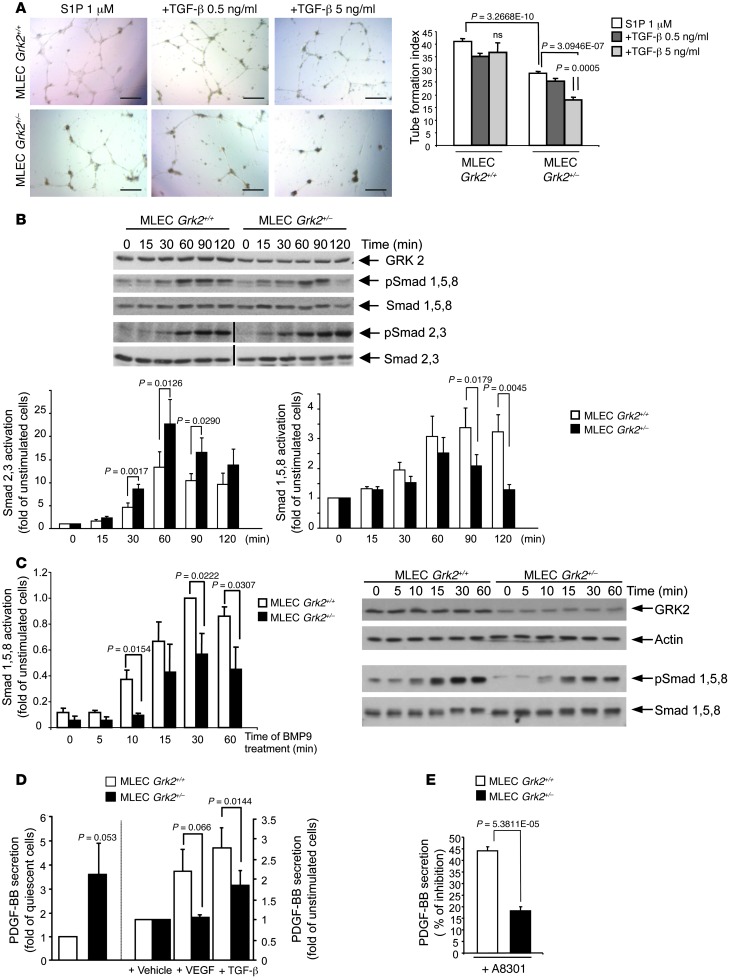
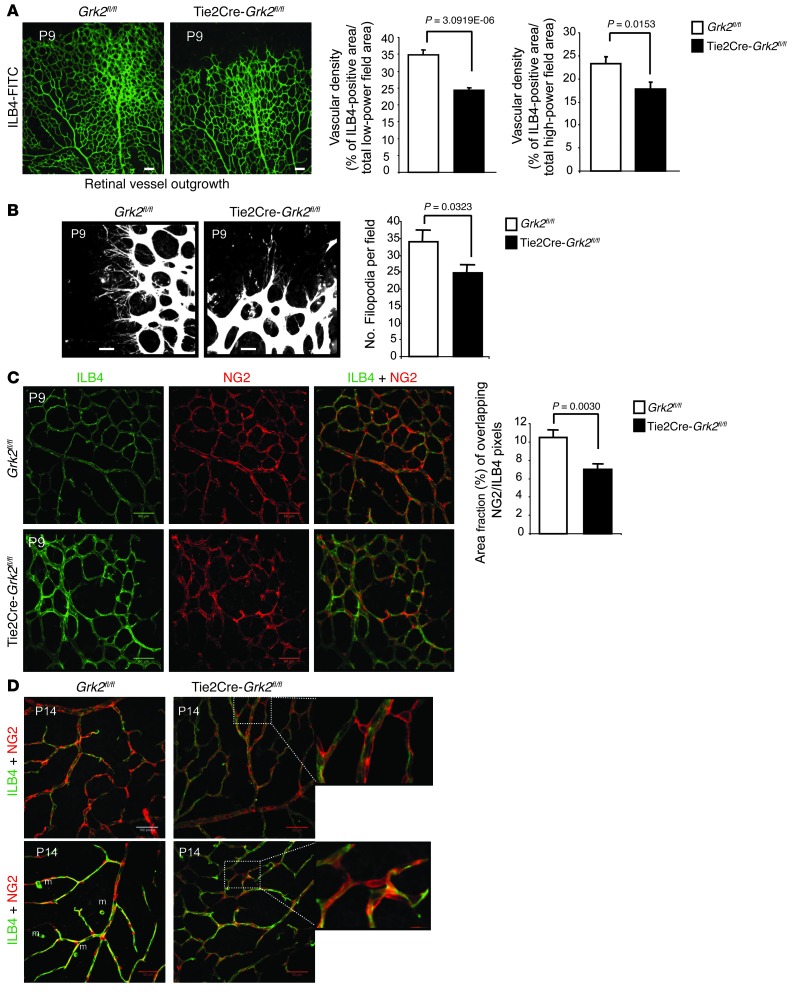
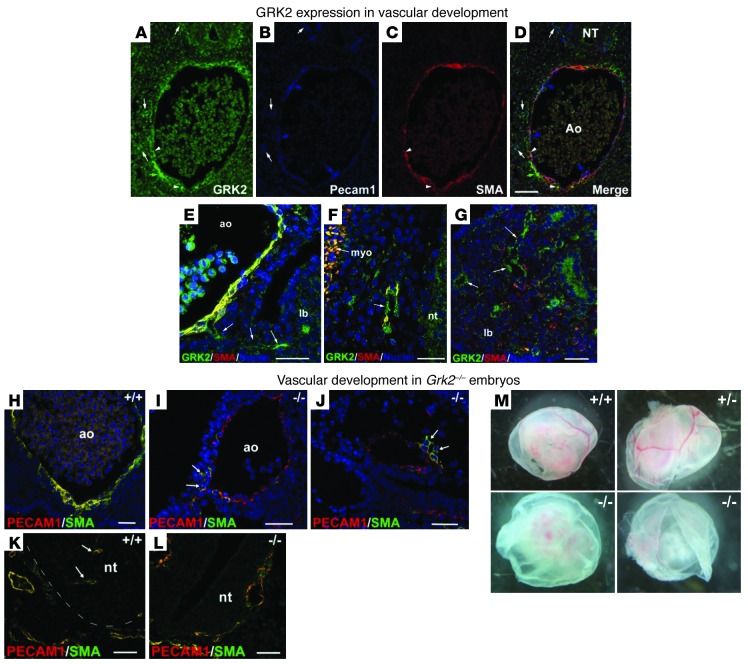
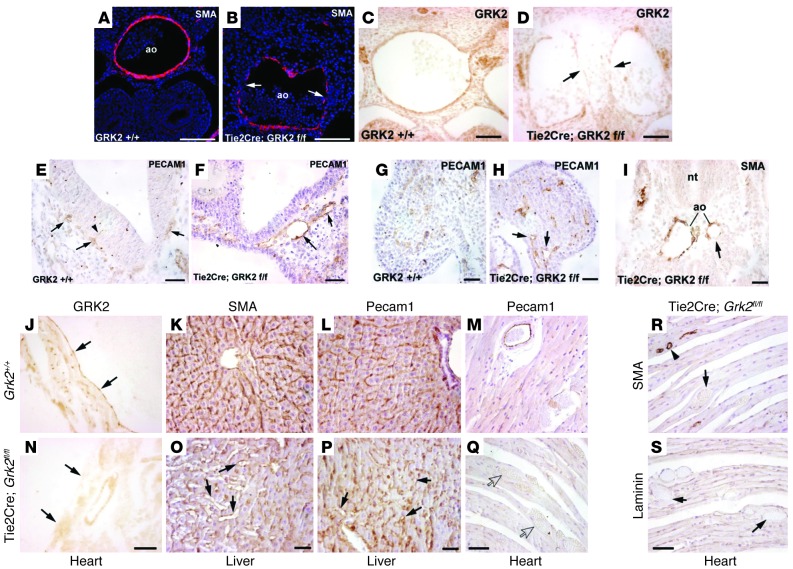
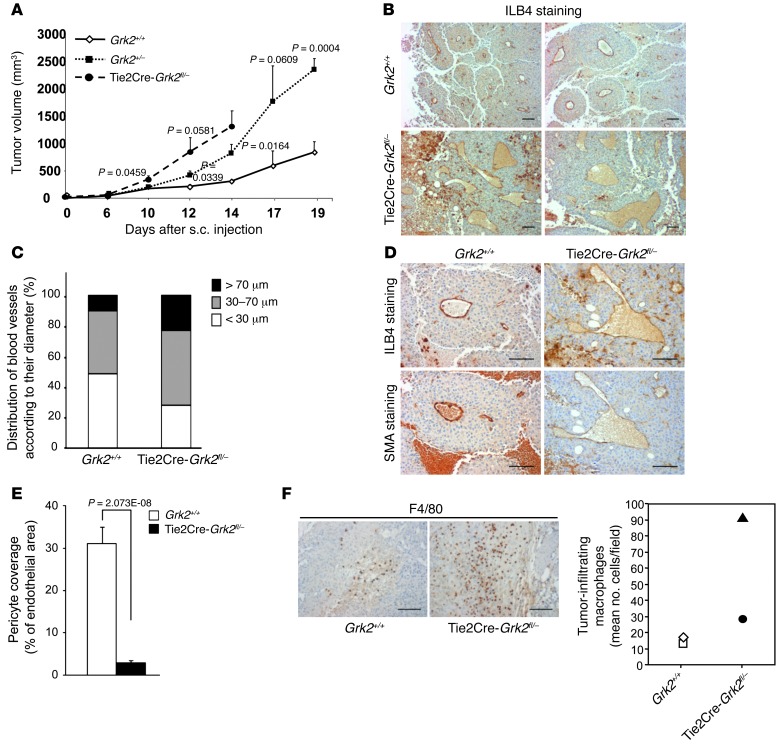
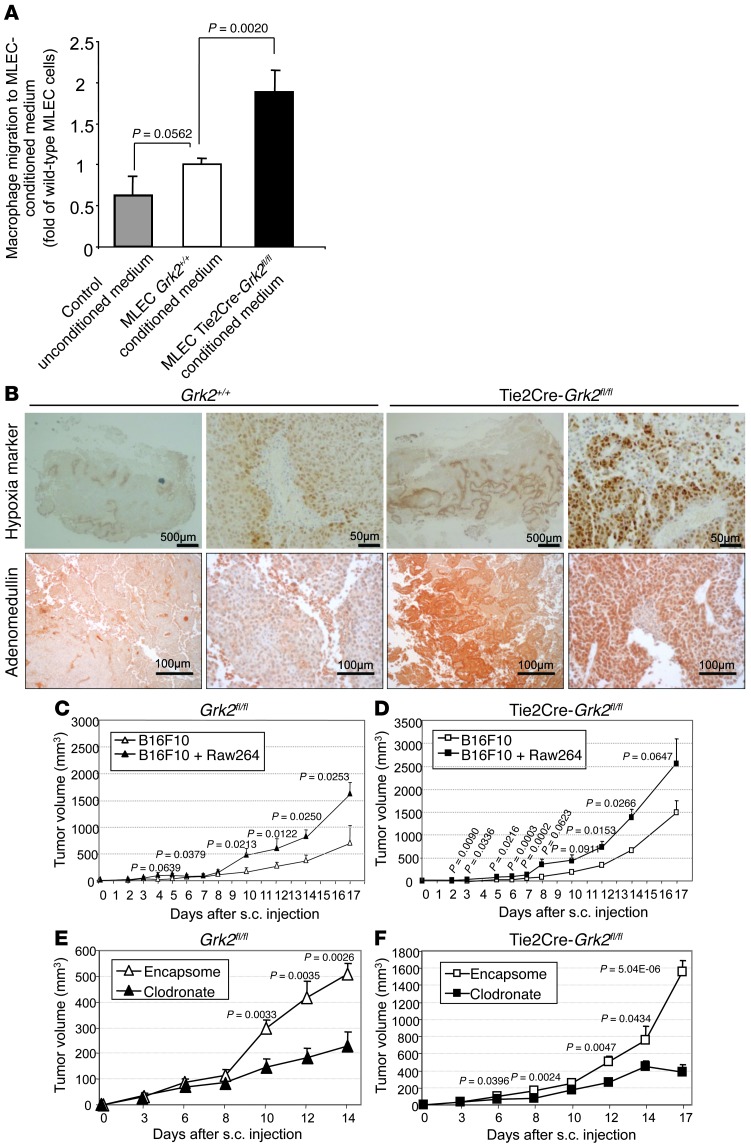
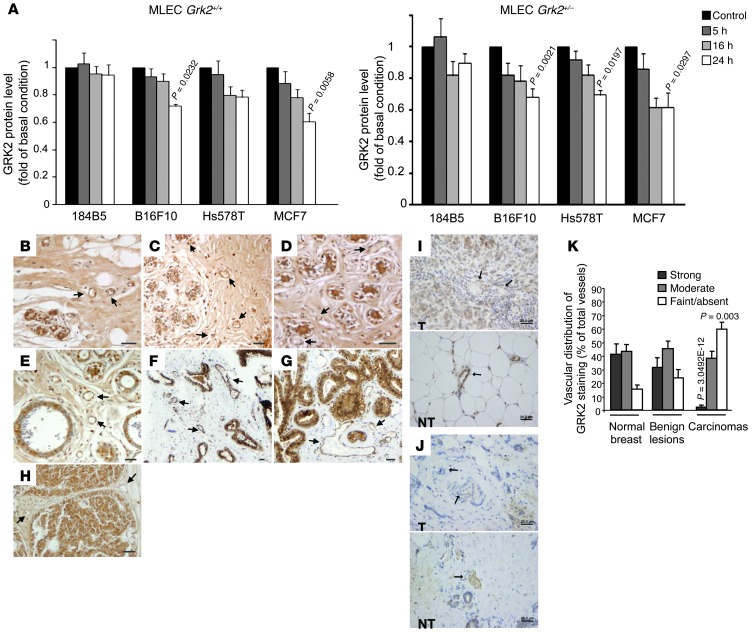
Tumor vessel dysfunction is a pivotal event in cancer progression. Using an in vivo neovascularization model, we identified G protein-coupled receptor kinase 2 (GRK2) as a key angiogenesis regulator. An impaired angiogenic response involving immature vessels was observed in mice hemizygous for Grk2 or in animals with endothelium-specific Grk2 silencing. ECs isolated from these animals displayed intrinsic alterations in migration, TGF-β signaling, and formation of tubular networks. Remarkably, an altered pattern of vessel growth and maturation was detected in postnatal retinas from endothelium-specific Grk2 knockout animals. Mouse embryos with systemic or endothelium-selective Grk2 ablation had marked vascular malformations involving impaired recruitment of mural cells. Moreover, decreased endothelial Grk2 dosage accelerated tumor growth in mice, along with reduced pericyte vessel coverage and enhanced macrophage infiltration, and this transformed environment promoted decreased GRK2 in ECs and human breast cancer vessels. Our study suggests that GRK2 downregulation is a relevant event in the tumoral angiogenic switch.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
