

AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function
- PMID: 24135141
- PMCID: PMC3809777
- DOI: 10.1172/JCI66218
AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function
Abstract
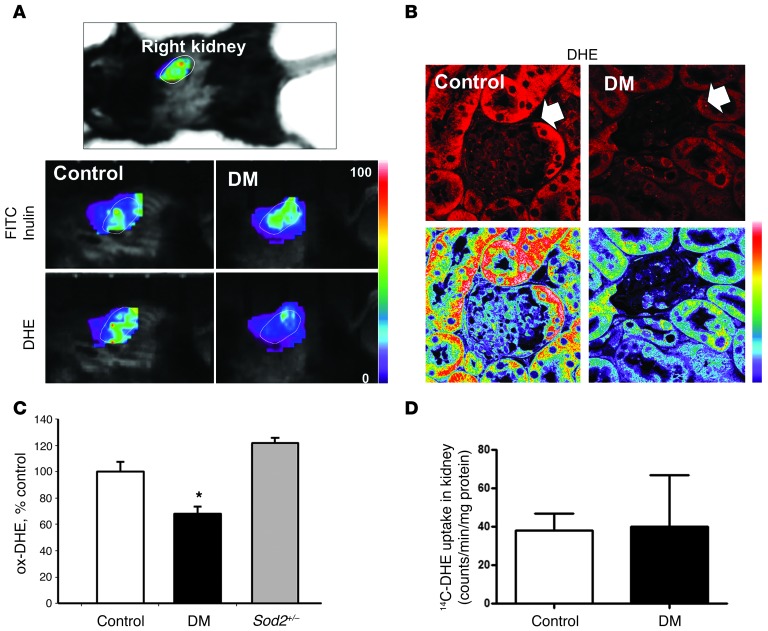
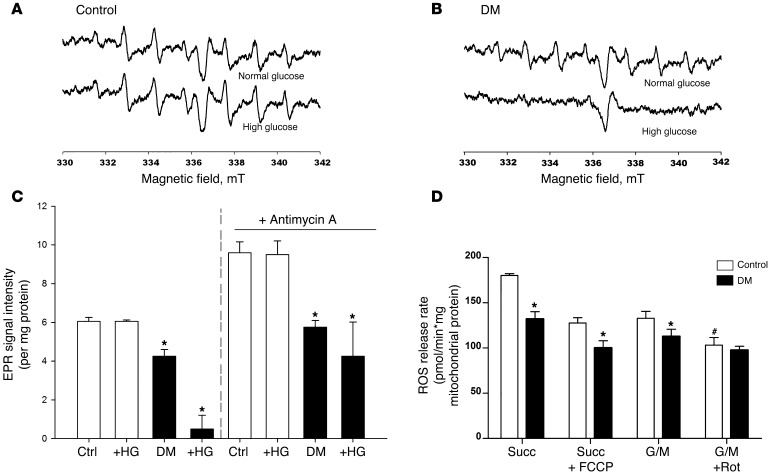
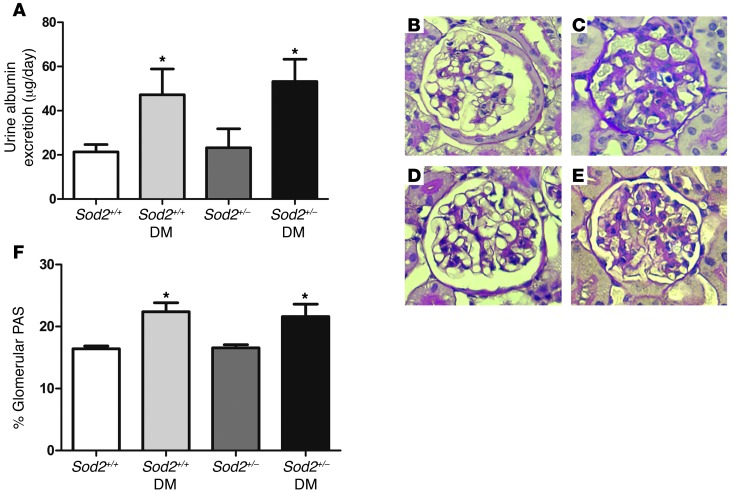
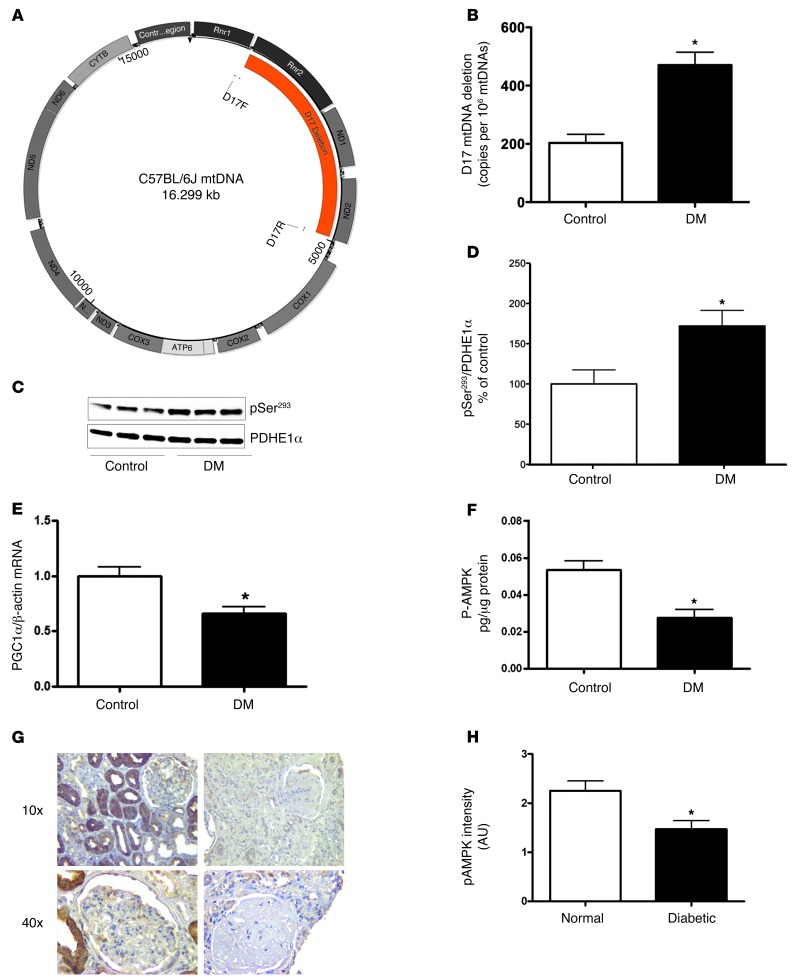
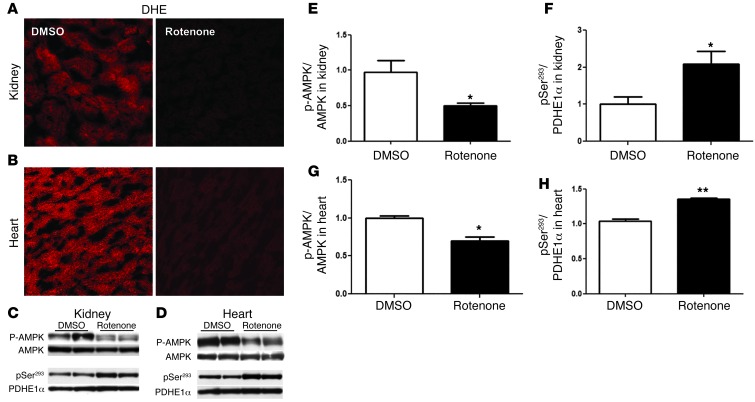
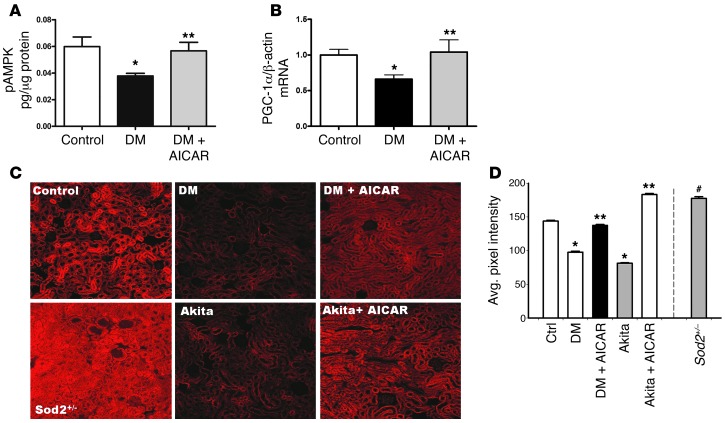
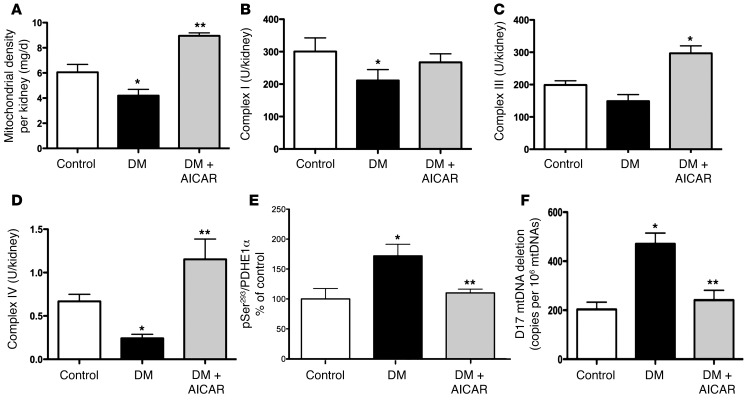
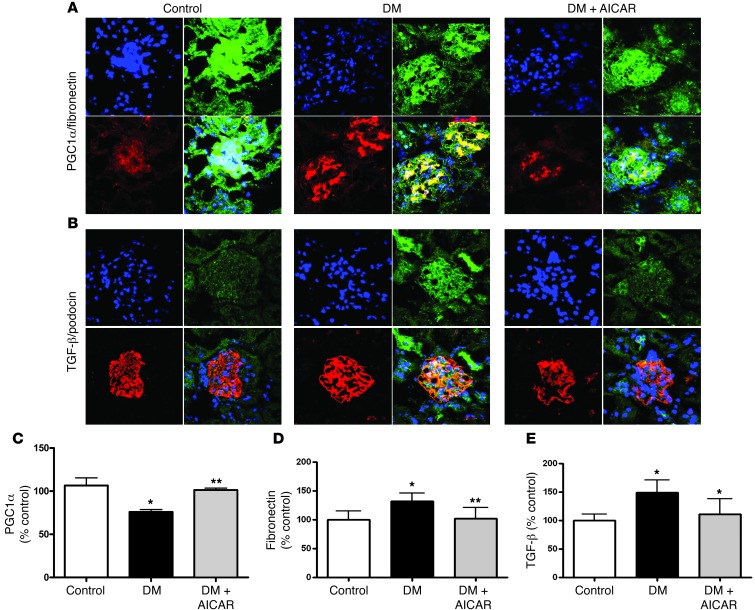
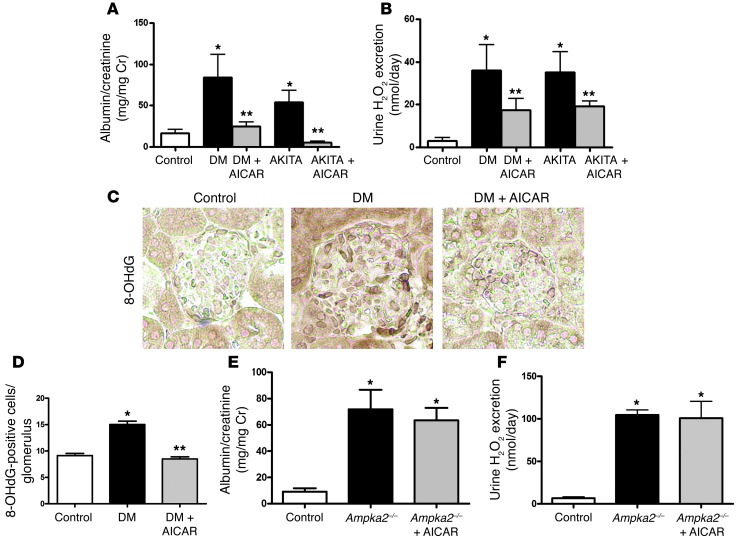
Diabetic microvascular complications have been considered to be mediated by a glucose-driven increase in mitochondrial superoxide anion production. Here, we report that superoxide production was reduced in the kidneys of a steptozotocin-induced mouse model of type 1 diabetes, as assessed by in vivo real-time transcutaneous fluorescence, confocal microscopy, and electron paramagnetic resonance analysis. Reduction of mitochondrial biogenesis and phosphorylation of pyruvate dehydrogenase (PDH) were observed in kidneys from diabetic mice. These observations were consistent with an overall reduction of mitochondrial glucose oxidation. Activity of AMPK, the major energy-sensing enzyme, was reduced in kidneys from both diabetic mice and humans. Mitochondrial biogenesis, PDH activity, and mitochondrial complex activity were rescued by treatment with the AMPK activator 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR). AICAR treatment induced superoxide production and was linked with glomerular matrix and albuminuria reduction in the diabetic kidney. Furthermore, diabetic heterozygous superoxide dismutase 2 (Sod2(+/-)) mice had no evidence of increased renal disease, and Ampka2(-/-) mice had increased albuminuria that was not reduced with AICAR treatment. Reduction of mitochondrial superoxide production with rotenone was sufficient to reduce AMPK phosphorylation in mouse kidneys. Taken together, these results demonstrate that diabetic kidneys have reduced superoxide and mitochondrial biogenesis and activation of AMPK enhances superoxide production and mitochondrial function while reducing disease activity.
Figures
Comment in
-
Mitochondrial ROS deficiency and diabetic complications: AMP[K]-lifying the adaptation to hyperglycemia.J Clin Invest. 2013 Nov;123(11):4573-6. doi: 10.1172/JCI72326. Epub 2013 Oct 25. J Clin Invest. 2013. PMID: 24135143 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
