

Genomics meets proteomics: identifying the culprits in disease
- PMID: 24135908
- PMCID: PMC4021166
- DOI: 10.1007/s00439-013-1376-2
Genomics meets proteomics: identifying the culprits in disease
Abstract
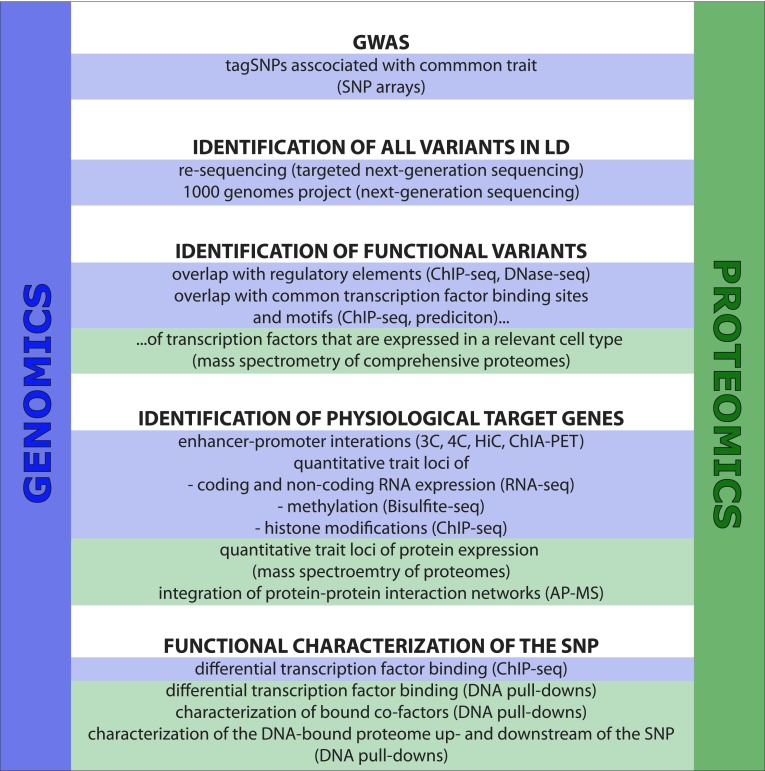
Genome-wide association studies (GWAS) revealed genomic risk loci that potentially have an impact on disease and phenotypic traits. This extensive resource holds great promise in providing novel directions for personalized medicine, including disease risk prediction, prevention and targeted medication. One of the major challenges that researchers face on the path between the initial identification of an association and precision treatment of patients is the comprehension of the biological mechanisms that underlie these associations. Currently, the focus to solve these questions lies on the integrative analysis of system-wide data on global genome variation, gene expression, transcription factor binding, epigenetic profiles and chromatin conformation. The generation of this data mainly relies on next-generation sequencing. However, due to multiple recent developments, mass spectrometry-based proteomics now offers additional, by the GWAS field so far hardly recognized possibilities for the identification of functional genome variants and, in particular, for the identification and characterization of (differentially) bound protein complexes as well as physiological target genes. In this review, we introduce these proteomics advances and suggest how they might be integrated in post-GWAS workflows. We argue that the combination of highly complementary techniques is powerful and can provide an unbiased, detailed picture of GWAS loci and their mechanistic involvement in disease.
Figures

References
-
- A Catalog of Published Genome-Wide Association Studies (2013). http://www.genome.gov/gwastudies. Accessed 11 May 2013
-
- Adams D, Altucci L, Antonarakis S, Ballesteros J, Beck S, Bird A, Bock C, Boehm B, Campo E, Caricasole A, Dahl F, Dermitzakis E, Enver T, Esteller M, Estivill X, Ferguson-Smith A, Fitzgibbon J, Flicek P, Giehl C, Graf T, Grosveld F, Guigo R, Gut I, Helin K, Jarvius J, Küppers R, Lehrach H, Lengauer T, Lernmark Å, Leslie D, Loeffler M, Macintyre E, Mai A, Martens J, Minucci S, Ouwehand W, Pelicci P, Pendeville H, Porse B, Rakyan V, Reik W, Schrappe M, Schübeler D, Seifert M, Siebert R, Simmons D, Soranzo N, Spicuglia S, Stratton M, Stunnenberg H, Tanay A, Torrents D, Valencia A, Vellenga E, Vingron M, Walter J, Willcocks S. BLUEPRINT to decode the epigenetic signature written in blood. Nat Biotechnol. 2012;30(3):224–226. doi: 10.1038/nbt.2153. - DOI - PubMed
-
- Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Bonnen PE, de Bakker PI, Deloukas P, Gabriel SB, Gwilliam R, Hunt S, Inouye M, Jia X, Palotie A, Parkin M, Whittaker P, Chang K, Hawes A, Lewis LR, Ren Y, Wheeler D, Muzny DM, Barnes C, Darvishi K, Hurles M, Korn JM, Kristiansson K, Lee C, McCarrol SA, Nemesh J, Keinan A, Montgomery SB, Pollack S, Price AL, Soranzo N, Gonzaga-Jauregui C, Anttila V, Brodeur W, Daly MJ, Leslie S, McVean G, Moutsianas L, Nguyen H, Zhang Q, Ghori MJ, McGinnis R, McLaren W, Takeuchi F, Grossman SR, Shlyakhter I, Hostetter EB, Sabeti PC, Adebamowo CA, Foster MW, Gordon DR, Licinio J, Manca MC, Marshall PA, Matsuda I, Ngare D, Wang VO, Reddy D, Rotimi CN, Royal CD, Sharp RR, Zeng C, Brooks LD, McEwen JE. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467(7311):52–58. doi: 10.1038/nature09298. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
