

Selective ribosome profiling as a tool for studying the interaction of chaperones and targeting factors with nascent polypeptide chains and ribosomes
- PMID: 24136347
- PMCID: PMC4001812
- DOI: 10.1038/nprot.2013.133
Selective ribosome profiling as a tool for studying the interaction of chaperones and targeting factors with nascent polypeptide chains and ribosomes
Abstract
A plethora of factors is involved in the maturation of newly synthesized proteins, including chaperones, membrane targeting factors and enzymes. Many factors act co-translationally through association with ribosome-nascent chain complexes (RNCs), but their target specificities and modes of action remain poorly understood. We developed selective ribosome profiling (SeRP) to identify substrate pools and points of RNC engagement of these factors. SeRP is based on sequencing mRNA fragments covered by translating ribosomes (general ribosome profiling (RP)), combined with a procedure to selectively isolate RNCs whose nascent polypeptides are associated with the factor of interest. Factor-RNC interactions are stabilized by cross-linking; the resulting factor-RNC adducts are nuclease-treated to generate monosomes, and then they are affinity purified. The ribosome-extracted mRNA footprints are converted to DNA libraries for deep sequencing. The protocol is specified for general RP and SeRP in bacteria. It was first applied to the chaperone trigger factor (TF) and is readily adaptable to other co-translationally acting factors, including eukaryotic factors. Factor-RNC purification and sequencing library preparation takes 7-8 d, and sequencing and data analysis can be completed in 5-6 d.
Figures

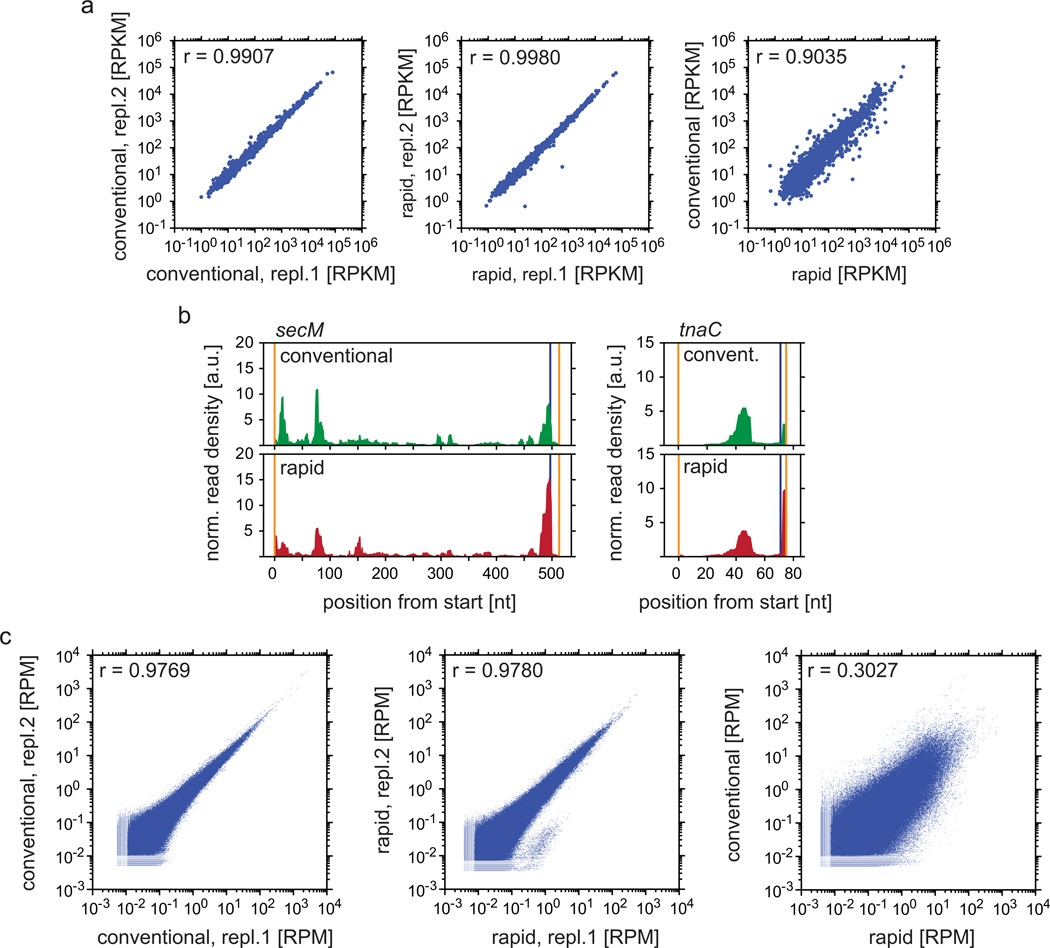
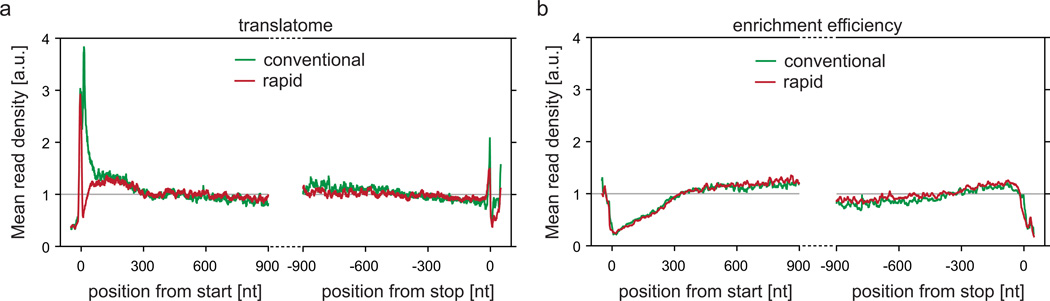
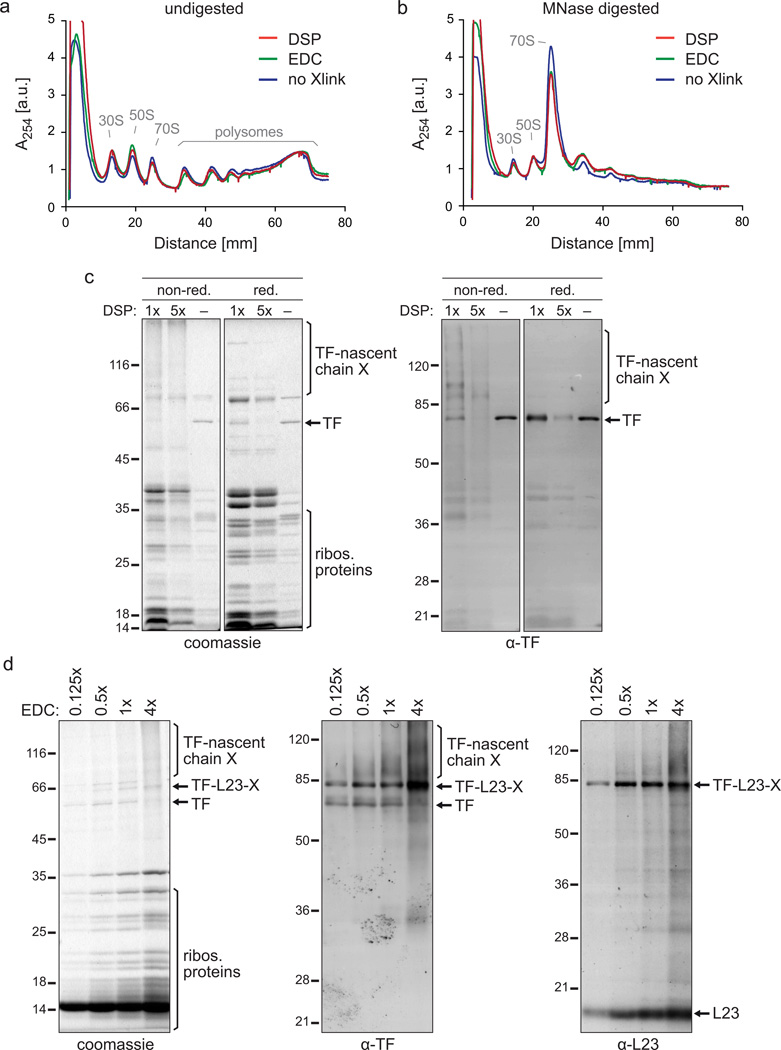
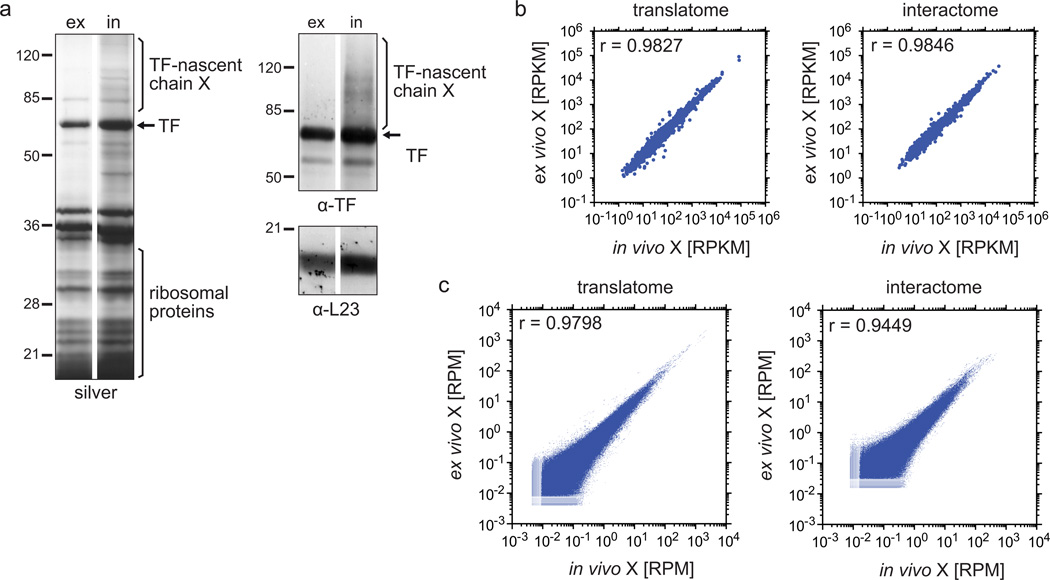

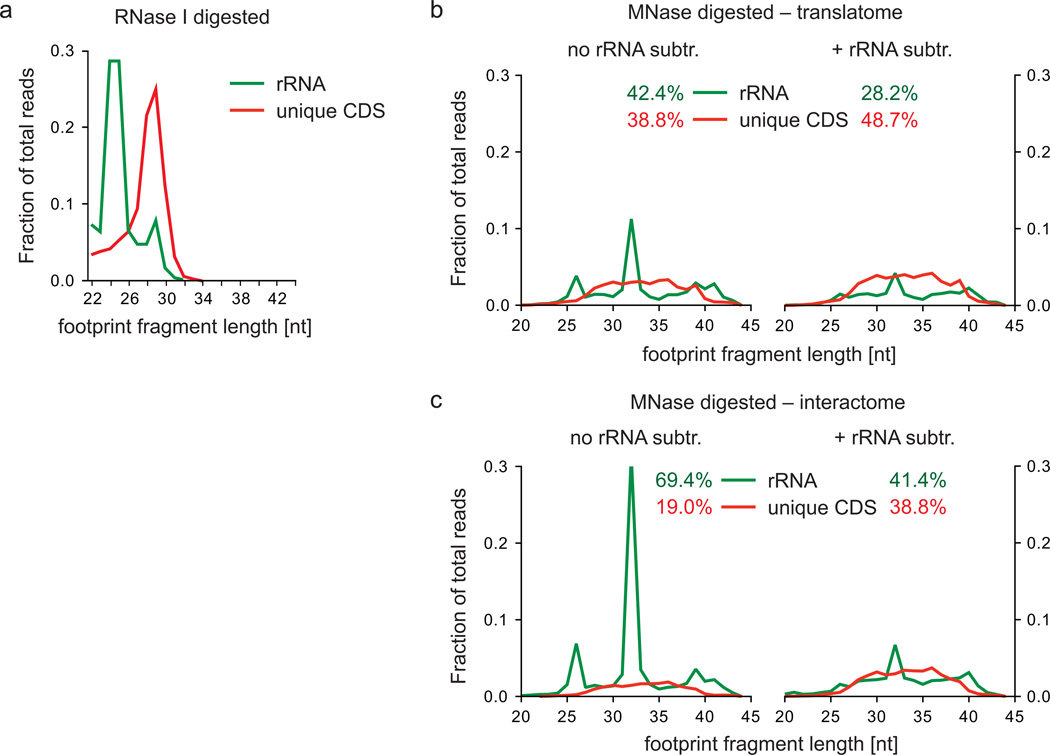
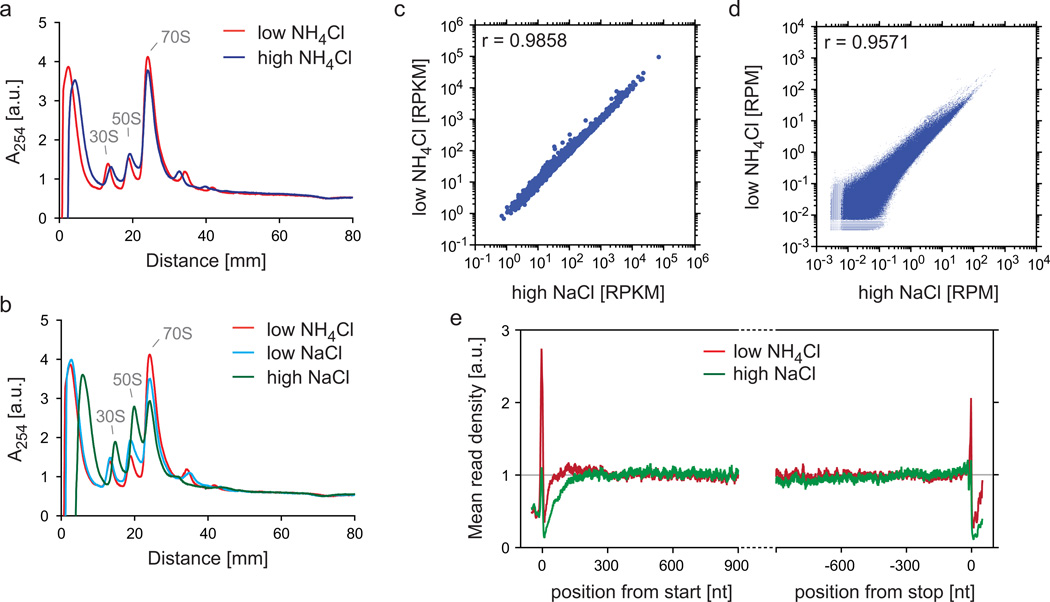
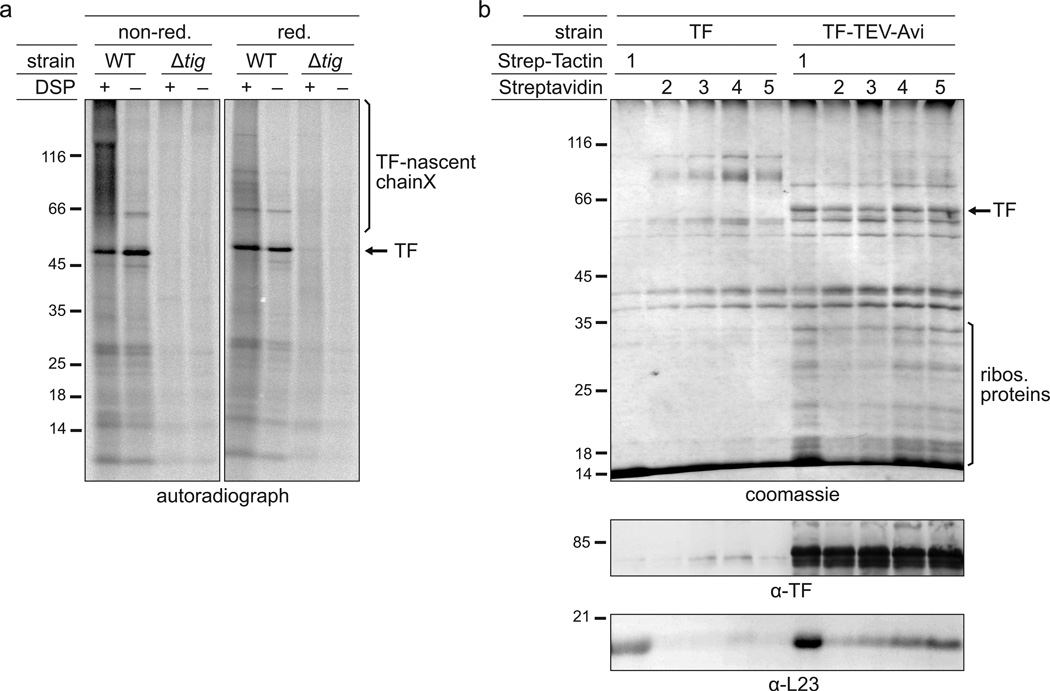
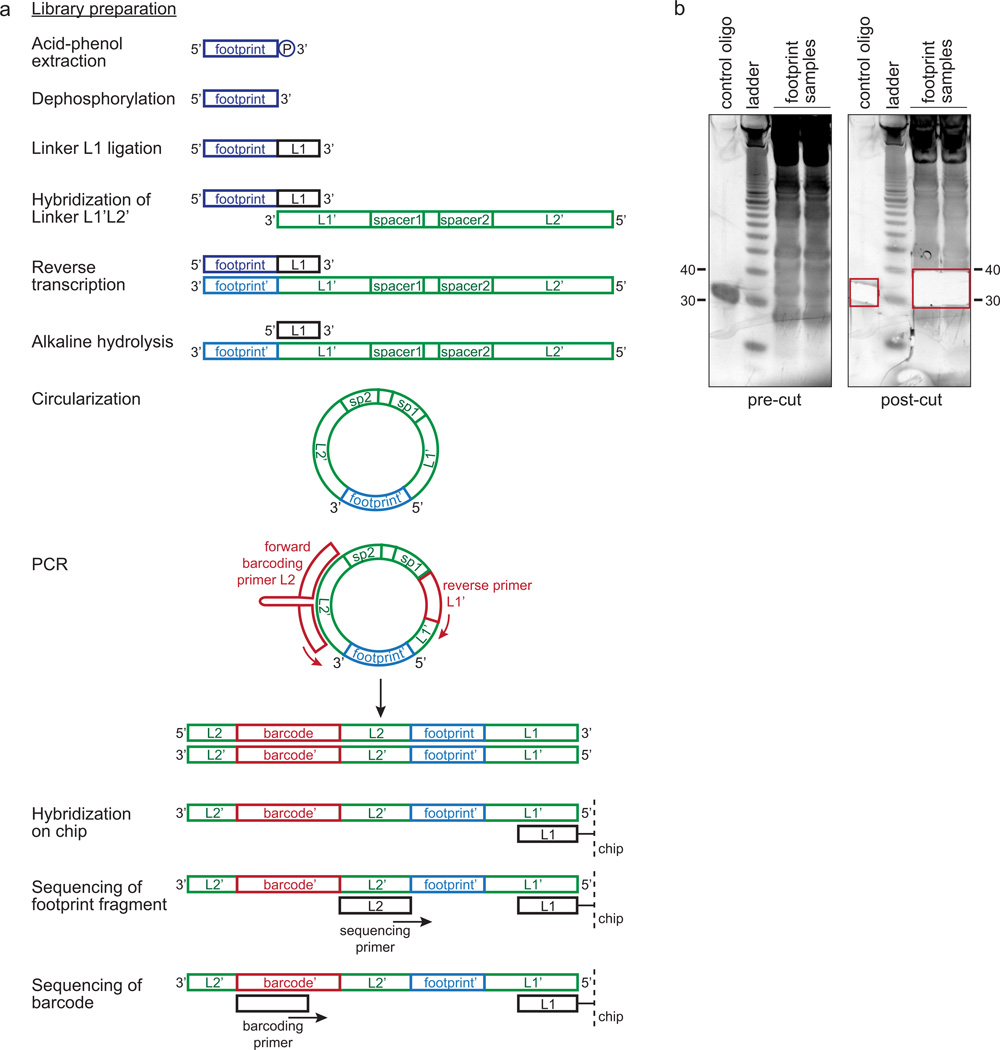
References
-
- Kramer G, Boehringer D, Ban N, Bukau B. The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat Struct Mol Biol. 2009;16:589–597. - PubMed
-
- Starheim KK, Gevaert K, Arnesen T. Protein N-terminal acetyltransferases: when the start matters. Trends Biochem Sci. 2012;37:152–161. - PubMed
-
- Jones JD, O'Connor CD. Protein acetylation in prokaryotes. Proteomics. 2011;11:3012–3022. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
