

Exonic duplication CNV of NDRG1 associated with autosomal-recessive HMSN-Lom/CMT4D
- PMID: 24136616
- PMCID: PMC4224029
- DOI: 10.1038/gim.2013.155
Exonic duplication CNV of NDRG1 associated with autosomal-recessive HMSN-Lom/CMT4D
Erratum in
- Genet Med. 2014 Feb;16(2):203
Abstract
Purpose: Copy-number variations as a mutational mechanism contribute significantly to human disease. Approximately one-half of the patients with Charcot-Marie-Tooth (CMT) disease have a 1.4 Mb duplication copy-number variation as the cause of their neuropathy. However, non-CMT1A neuropathy patients rarely have causative copy-number variations, and to date, autosomal-recessive disease has not been associated with copy-number variation as a mutational mechanism.
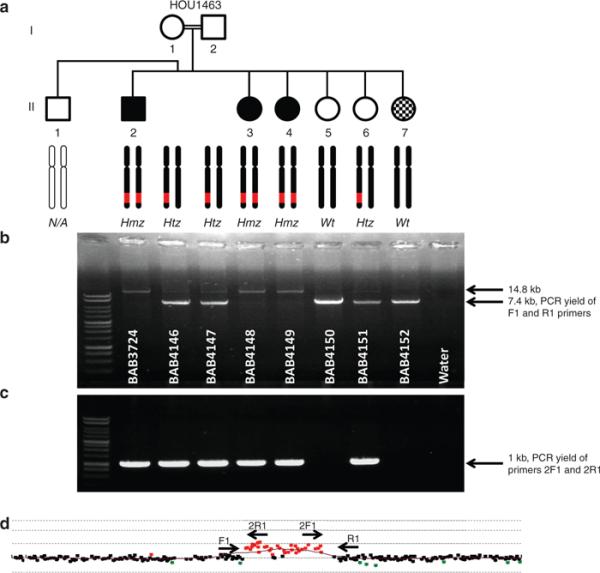
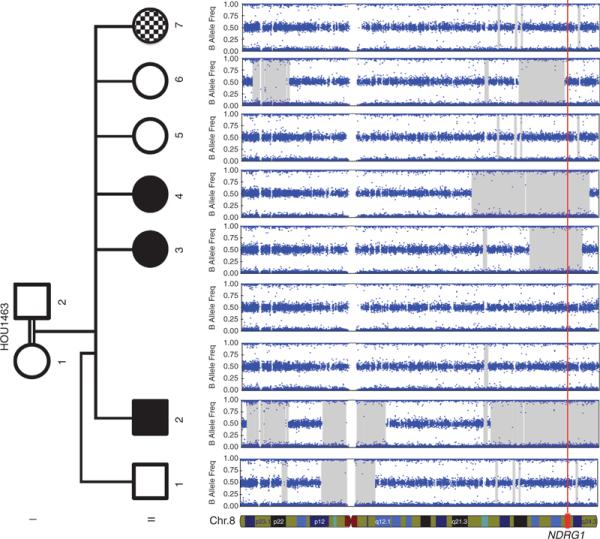
Methods: We performed Agilent 8 × 60 K array comparative genomic hybridization on DNA from 12 recessive Turkish families with CMT disease. Additional molecular studies were conducted to detect breakpoint junctions and to evaluate gene expression levels in a family in which we detected an intragenic duplication copy-number variation.
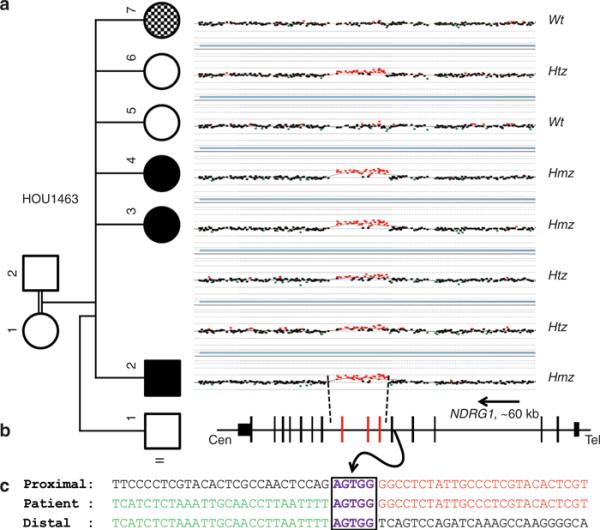
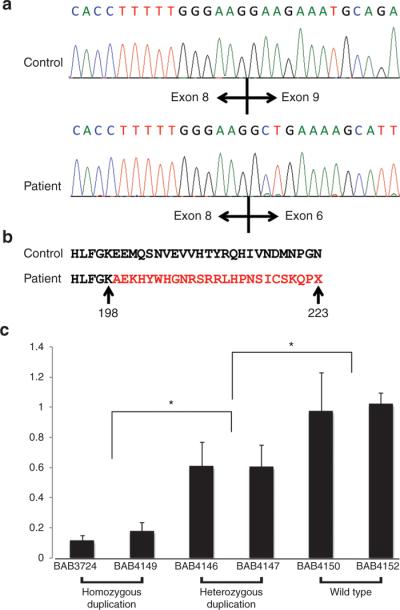
Results: We detected an ~6.25 kb homozygous intragenic duplication in NDRG1, a gene known to be causative for recessive HMSNL/CMT4D, in three individuals from a Turkish family with CMT neuropathy. Further studies showed that this intragenic copy-number variation resulted in a homozygous duplication of exons 6-8 that caused decreased mRNA expression of NDRG1.
Conclusion: Exon-focused high-resolution array comparative genomic hybridization enables the detection of copy-number variation carrier states in recessive genes, particularly small copy-number variations encompassing or disrupting single genes. In families for whom a molecular diagnosis has not been elucidated by conventional clinical assays, an assessment for copy-number variations in known CMT genes might be considered.
Figures
Similar articles
-
A novel NDRG1 mutation in a non-Romani patient with CMT4D/HMSN-Lom.J Peripher Nerv Syst. 2017 Mar;22(1):47-50. doi: 10.1111/jns.12201. J Peripher Nerv Syst. 2017. PMID: 27982524
-
Identification and functional characterization of two missense mutations in NDRG1 associated with Charcot-Marie-Tooth disease type 4D.Hum Mutat. 2017 Nov;38(11):1569-1578. doi: 10.1002/humu.23309. Epub 2017 Aug 23. Hum Mutat. 2017. PMID: 28776325
-
CMT4D (NDRG1 mutation): genotype-phenotype correlations.J Peripher Nerv Syst. 2013 Sep;18(3):261-5. doi: 10.1111/jns5.12039. J Peripher Nerv Syst. 2013. PMID: 24028195
-
[Molecular genetics of inherited neuropathies].Rinsho Shinkeigaku. 2006 Jan;46(1):1-18. Rinsho Shinkeigaku. 2006. PMID: 16541790 Review. Japanese.
-
Charcot-Marie-Tooth disease and related inherited neuropathies.Medicine (Baltimore). 1996 Sep;75(5):233-50. doi: 10.1097/00005792-199609000-00001. Medicine (Baltimore). 1996. PMID: 8862346 Review.
Cited by
-
A Review of Copy Number Variants in Inherited Neuropathies.Curr Genomics. 2018 Sep;19(6):412-419. doi: 10.2174/1389202919666180330153316. Curr Genomics. 2018. PMID: 30258273 Free PMC article. Review.
-
Copy number variant and runs of homozygosity detection by microarrays enabled more precise molecular diagnoses in 11,020 clinical exome cases.Genome Med. 2019 May 17;11(1):30. doi: 10.1186/s13073-019-0639-5. Genome Med. 2019. PMID: 31101064 Free PMC article.
-
The role of combined SNV and CNV burden in patients with distal symmetric polyneuropathy.Genet Med. 2016 May;18(5):443-51. doi: 10.1038/gim.2015.124. Epub 2015 Sep 17. Genet Med. 2016. PMID: 26378787 Free PMC article.
-
HMSN Lom in 12 Czech patients, with one unusual case due to uniparental isodisomy of chromosome 8.J Hum Genet. 2017 Mar;62(3):431-435. doi: 10.1038/jhg.2016.148. Epub 2016 Dec 22. J Hum Genet. 2017. PMID: 28003645
-
Identification of novel pathogenic copy number variations in Charcot-Marie-Tooth disease.J Hum Genet. 2020 Mar;65(3):313-323. doi: 10.1038/s10038-019-0710-5. Epub 2019 Dec 18. J Hum Genet. 2020. PMID: 31852984
References
-
- England JD, Gronseth GS, Franklin G, et al. American Academy of Neurology. American Association of Neuromuscular and Electrodiagnostic Medicine. American Academy of Physical Medicine and Rehabilitation Practice parameter: the evaluation of distal symmetric polyneuropathy: the role of laboratory and genetic testing (an evidence-based review). Report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. PM R. 2009;1:5–13. - PubMed
-
- Wiszniewski W, Szigeti K, Lupski JR. Hereditary Motor and Sensory Neuropathies. 6th Vol. 3. Elsevier; San Francisco, CA: 2013.
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
