

Hypermutation of the inactive X chromosome is a frequent event in cancer
- PMID: 24139898
- PMCID: PMC3898475
- DOI: 10.1016/j.cell.2013.09.042
Hypermutation of the inactive X chromosome is a frequent event in cancer
Abstract
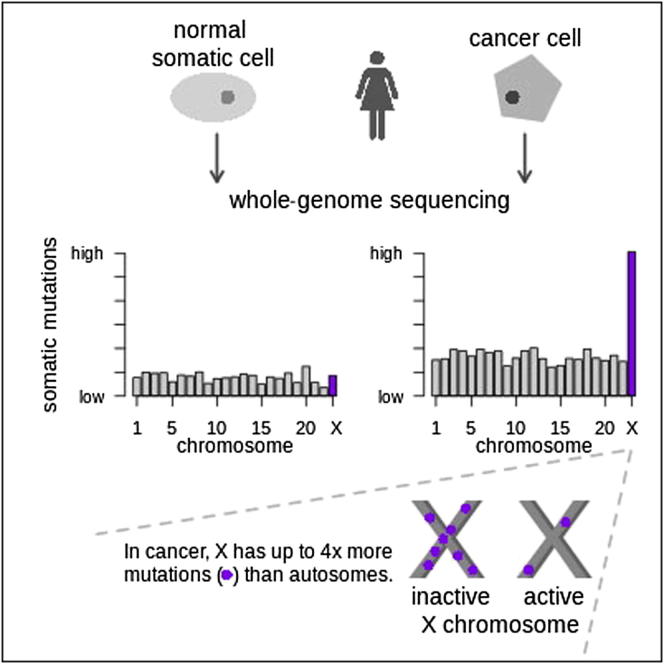
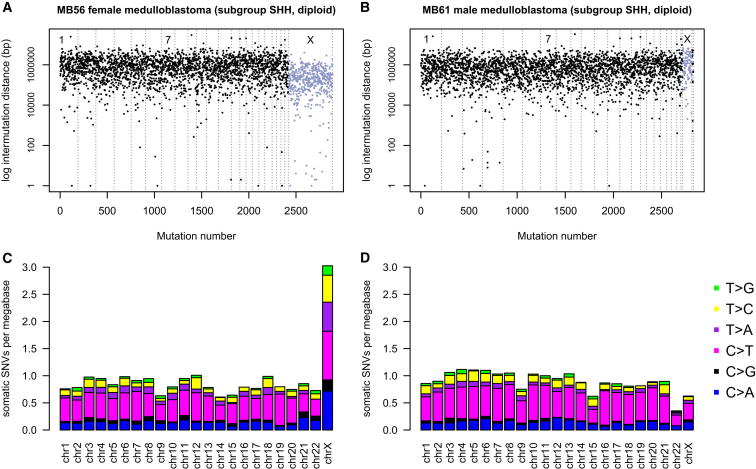
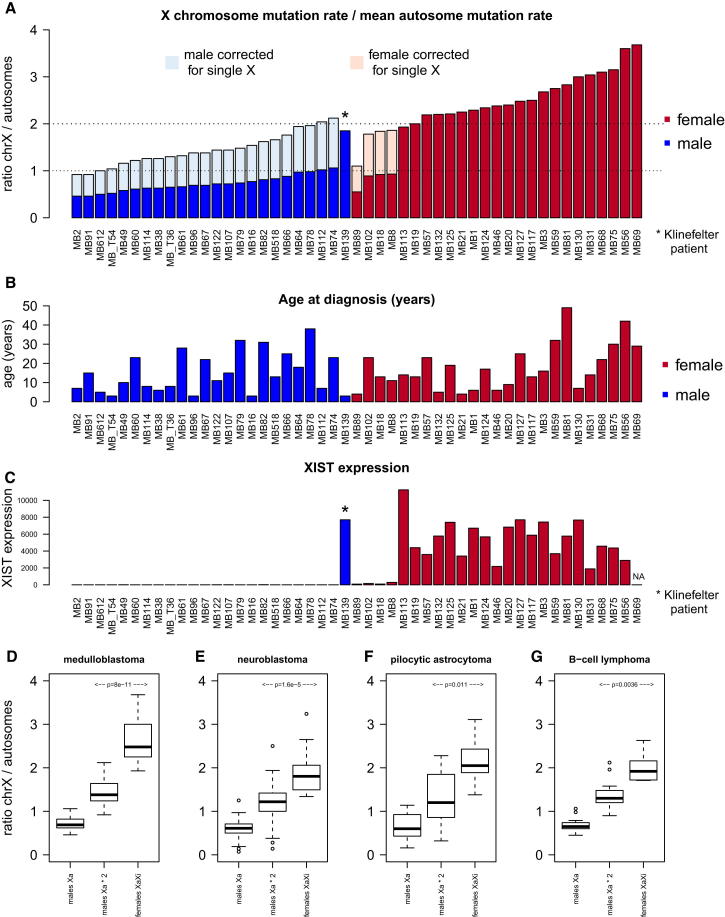
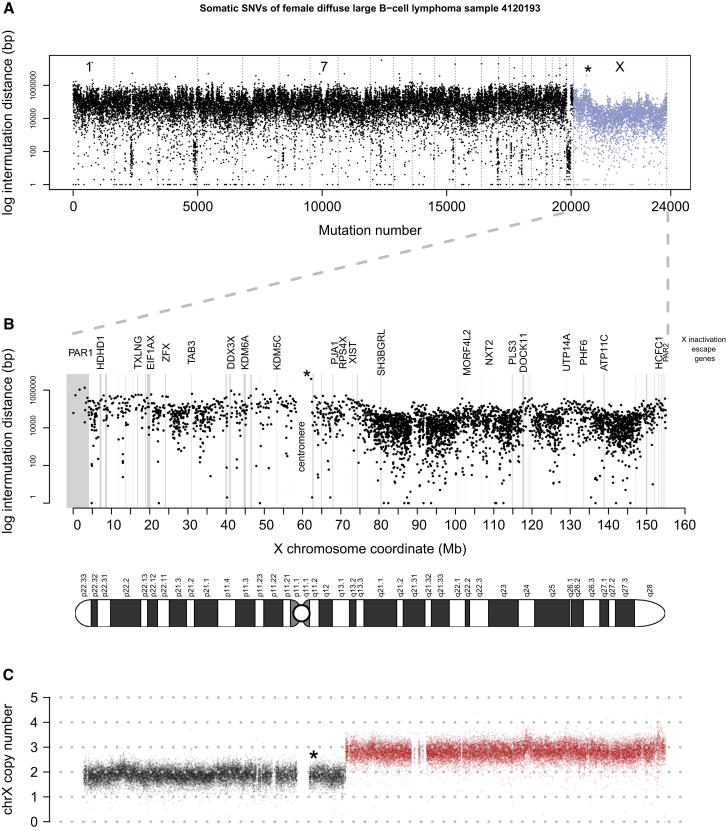
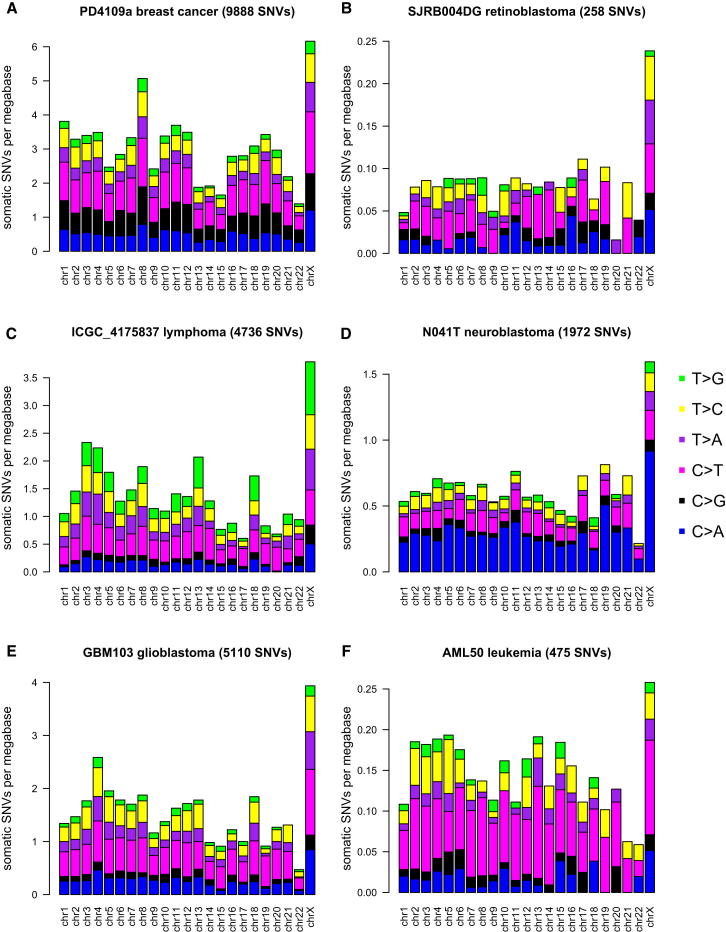
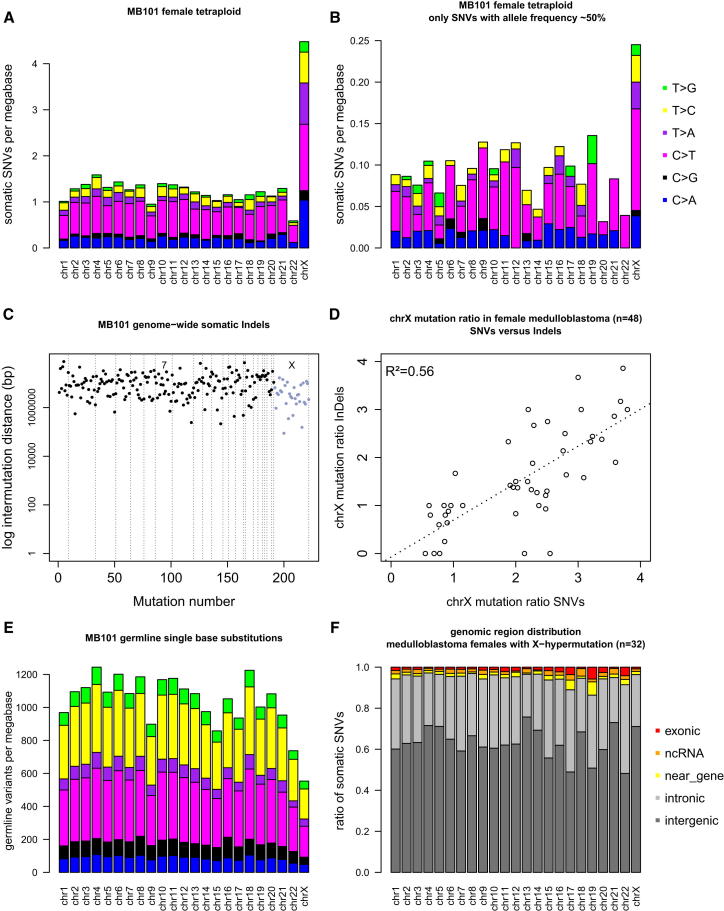
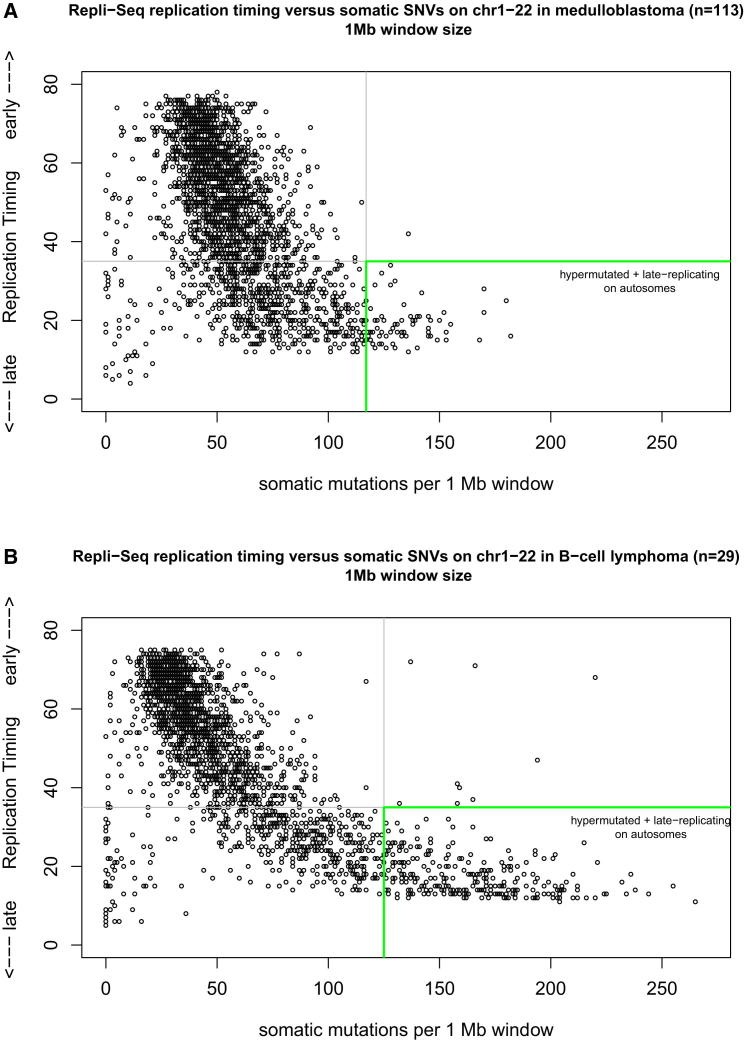
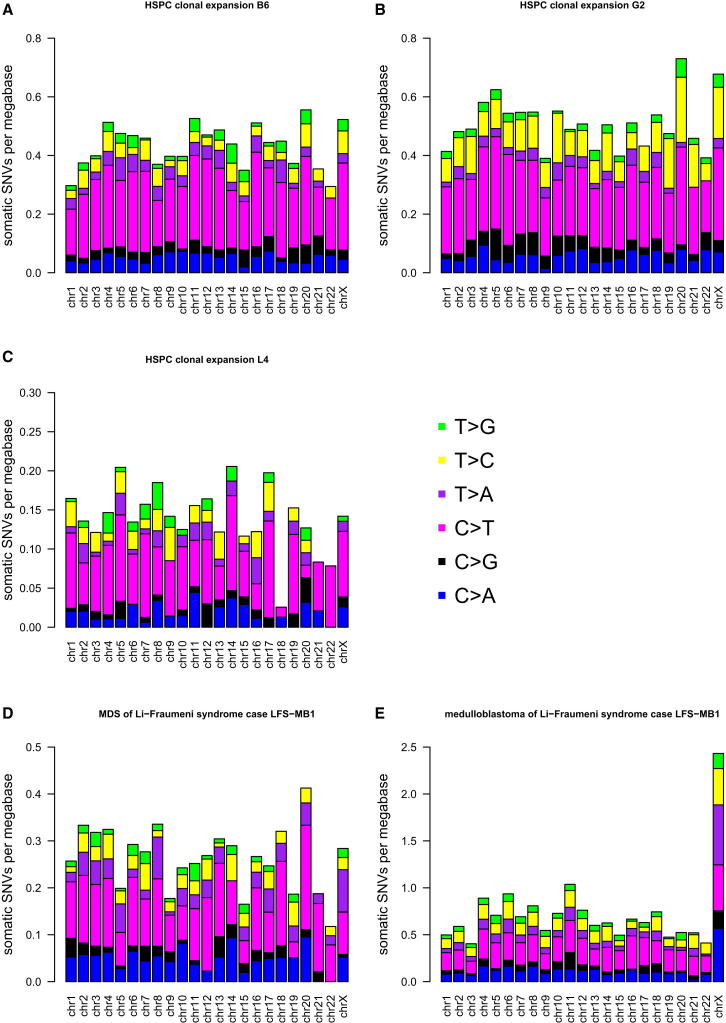
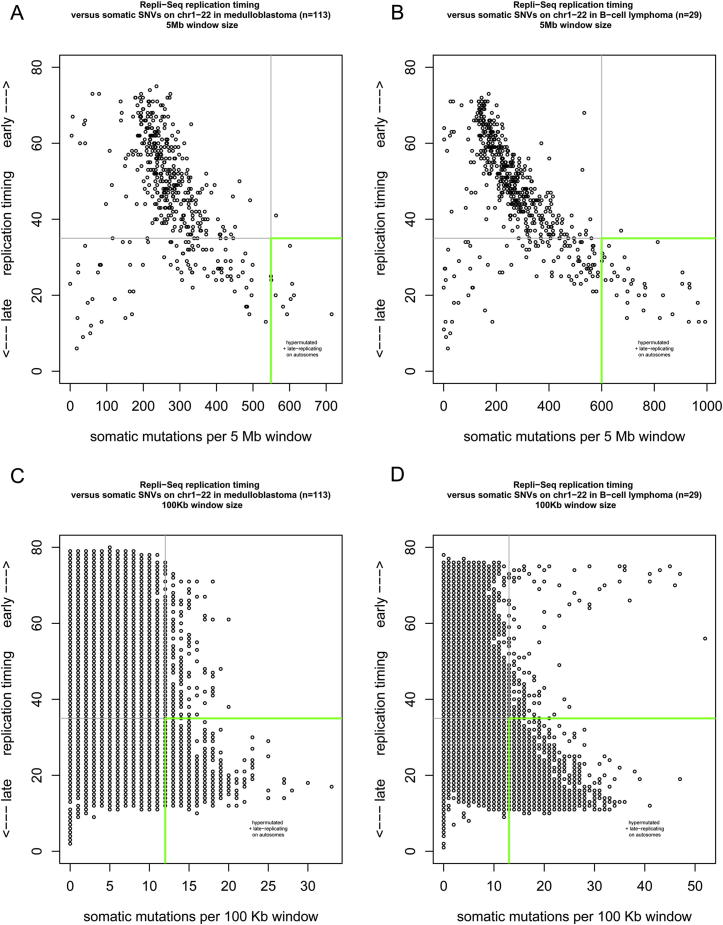
Mutation is a fundamental process in tumorigenesis. However, the degree to which the rate of somatic mutation varies across the human genome and the mechanistic basis underlying this variation remain to be fully elucidated. Here, we performed a cross-cancer comparison of 402 whole genomes comprising a diverse set of childhood and adult tumors, including both solid and hematopoietic malignancies. Surprisingly, we found that the inactive X chromosome of many female cancer genomes accumulates on average twice and up to four times as many somatic mutations per megabase, as compared to the individual autosomes. Whole-genome sequencing of clonally expanded hematopoietic stem/progenitor cells (HSPCs) from healthy individuals and a premalignant myelodysplastic syndrome (MDS) sample revealed no X chromosome hypermutation. Our data suggest that hypermutation of the inactive X chromosome is an early and frequent feature of tumorigenesis resulting from DNA replication stress in aberrantly proliferating cells.
Copyright © 2013 Elsevier Inc. All rights reserved.
Figures
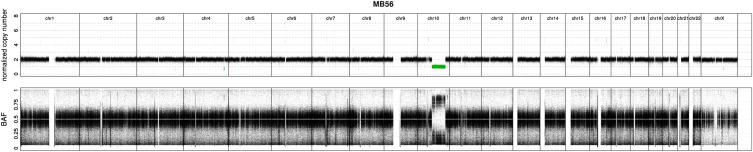
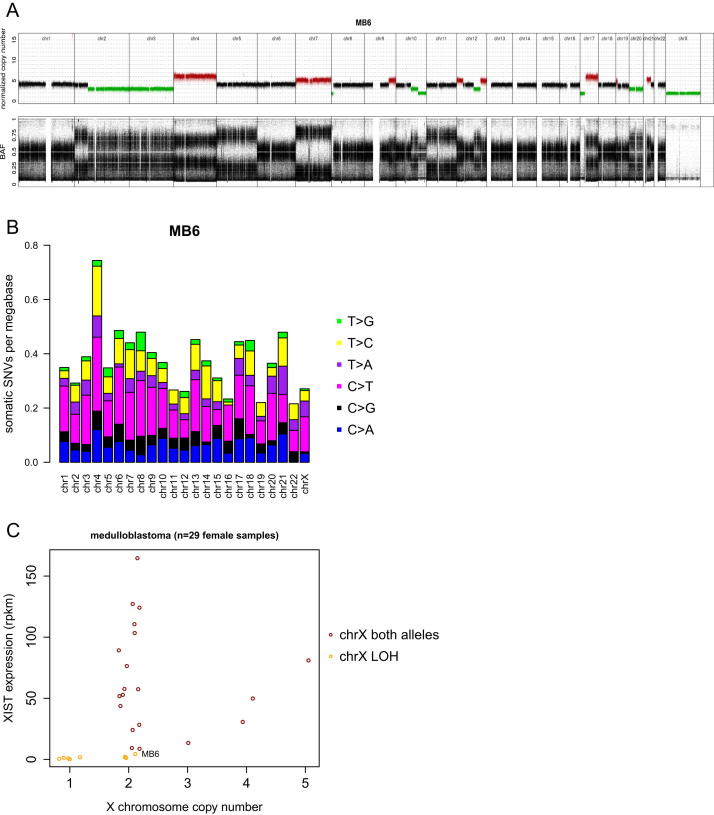
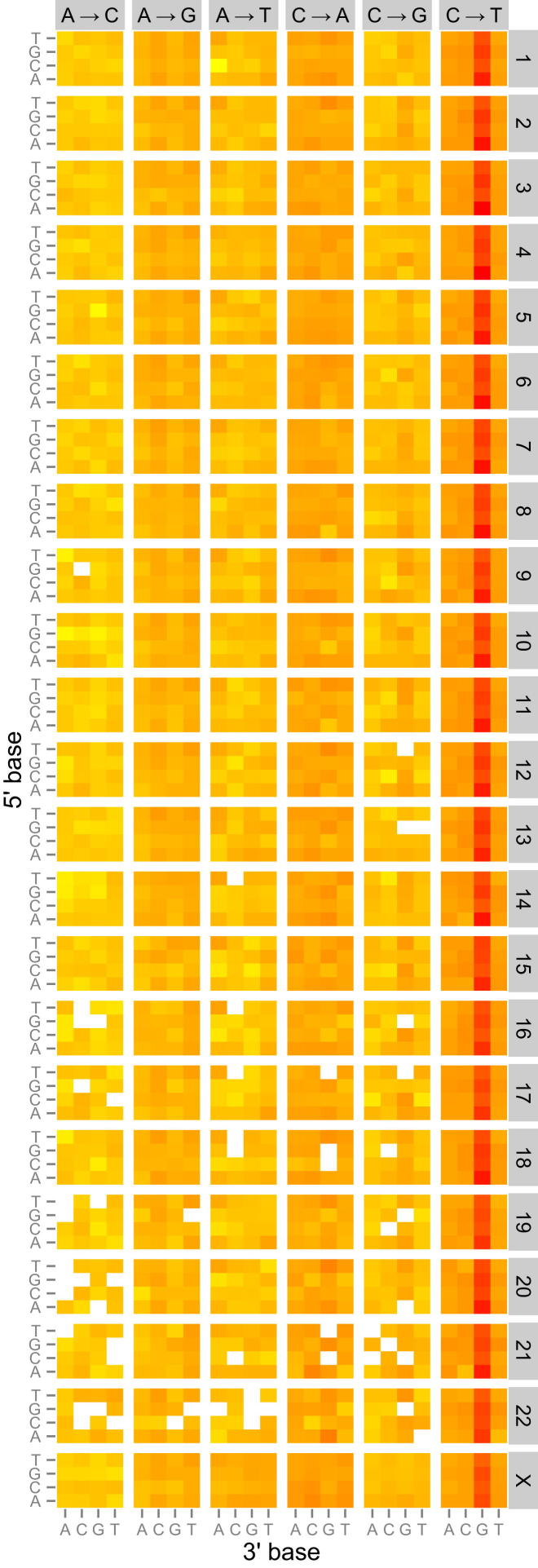
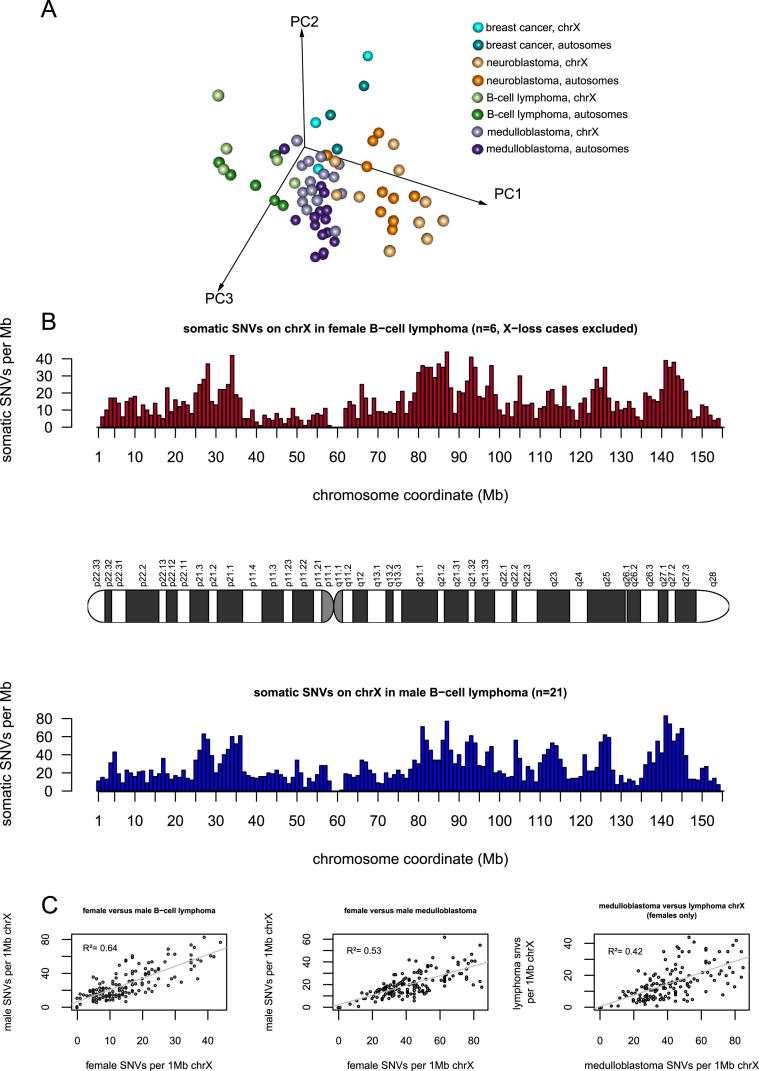
References
-
- Barakat T.S., Gribnau J. X chromosome inactivation in the cycle of life. Development. 2012;139:2085–2089. - PubMed
-
- Brown C.J., Hendrich B.D., Rupert J.L., Lafrenière R.G., Xing Y., Lawrence J., Willard H.F. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell. 1992;71:527–542. - PubMed
Supplemental References
-
- Wang Q., Gu L., Adey A., Radlwimmer B., Wang W., Hovestadt V., Bähr M., Wolf S., Shendure J., Eils R. Tagmentation-based whole-genome bisulfite sequencing. Nat. Protoc. 2013;8:2022–2032. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
