

mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc
- PMID: 24140020
- PMCID: PMC3840163
- DOI: 10.1016/j.cmet.2013.09.013
mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc
Abstract
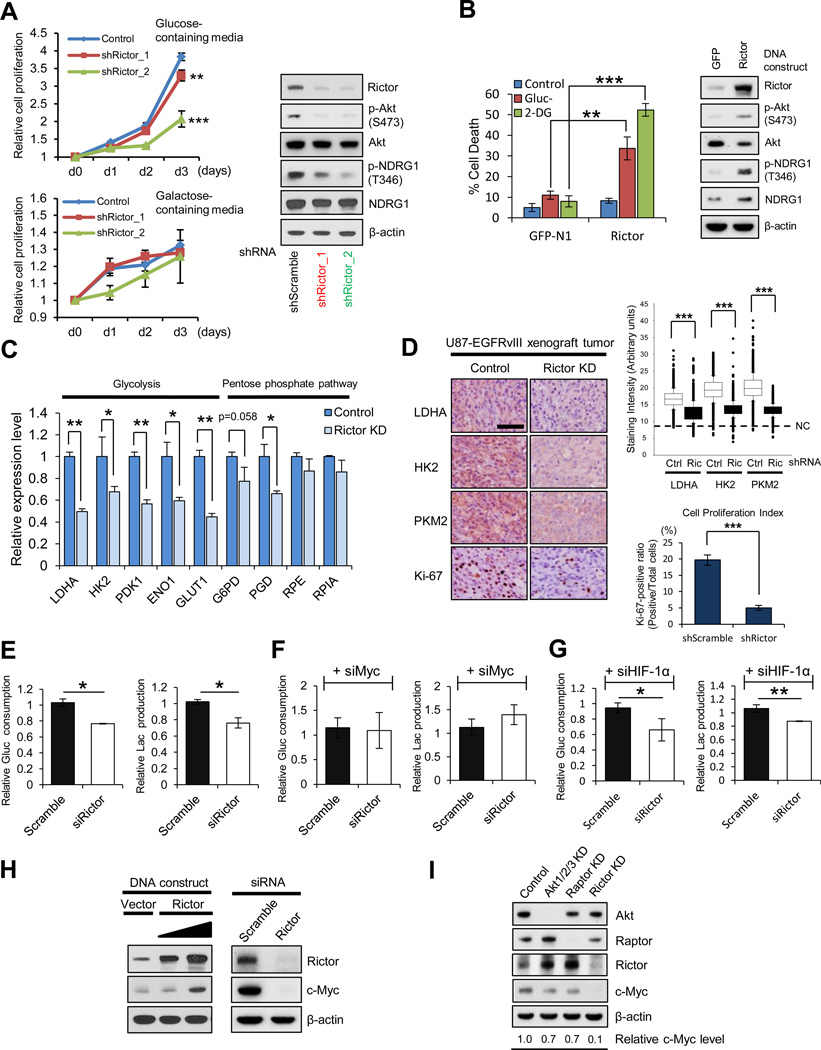
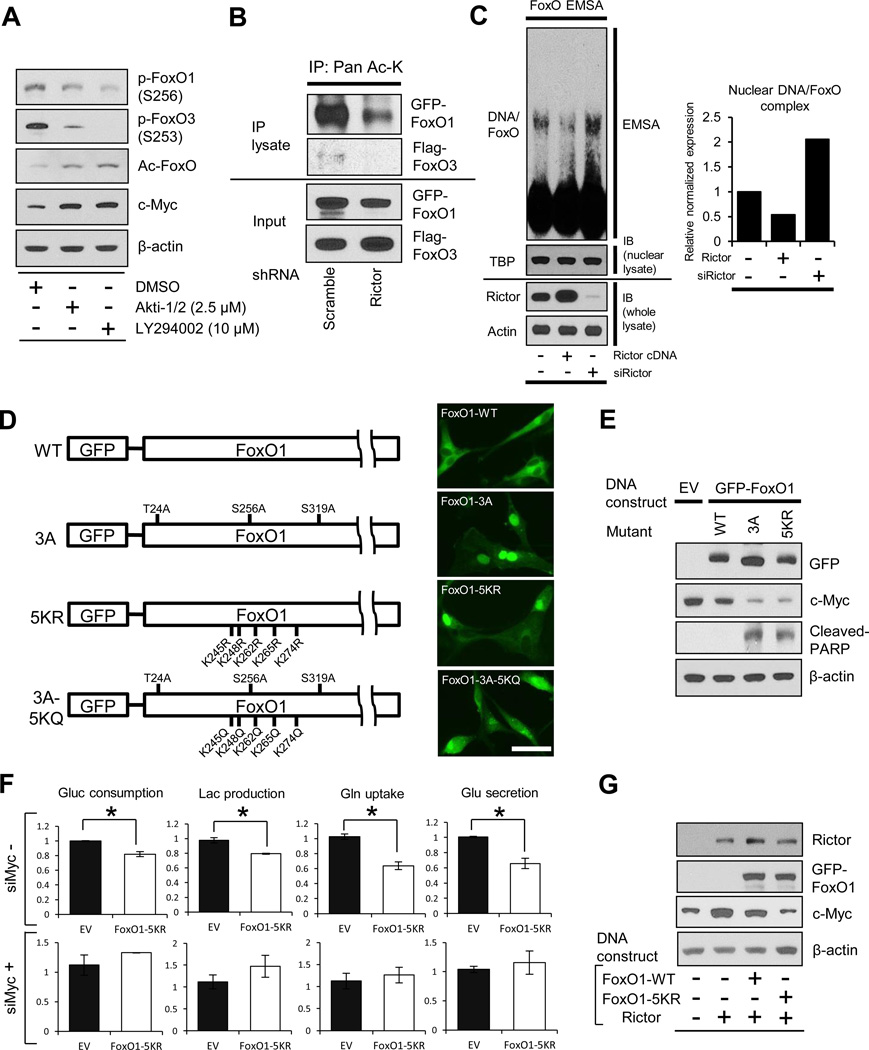
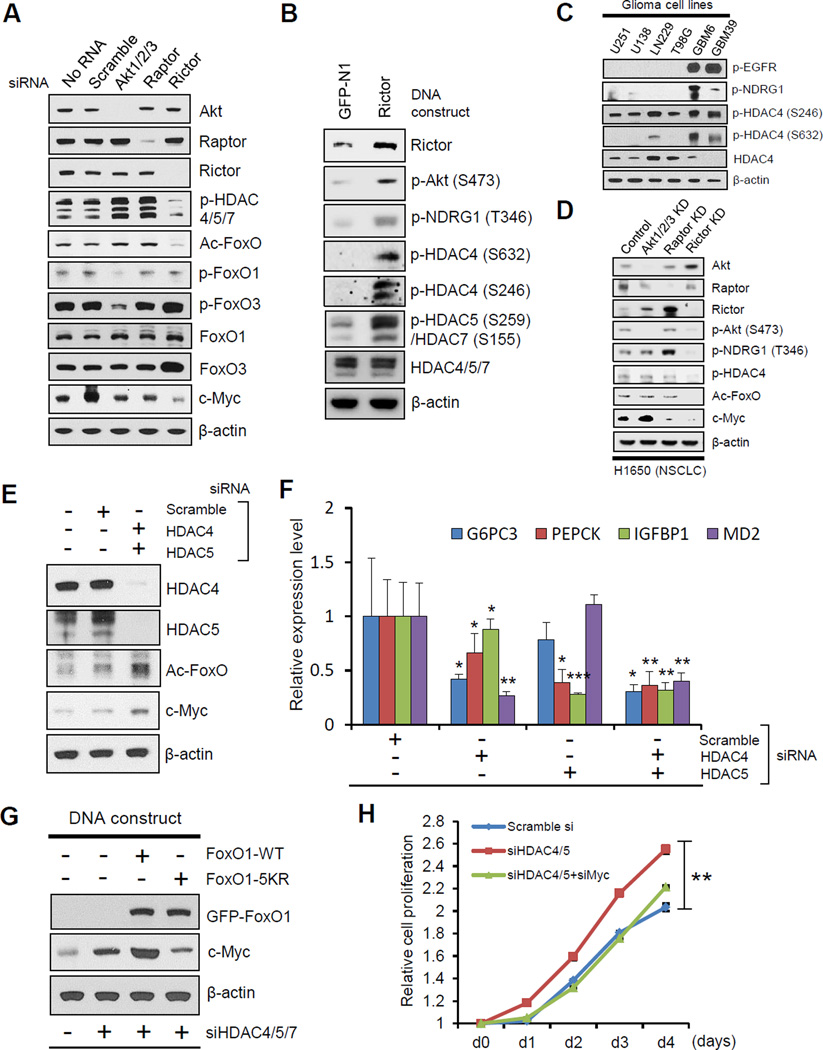
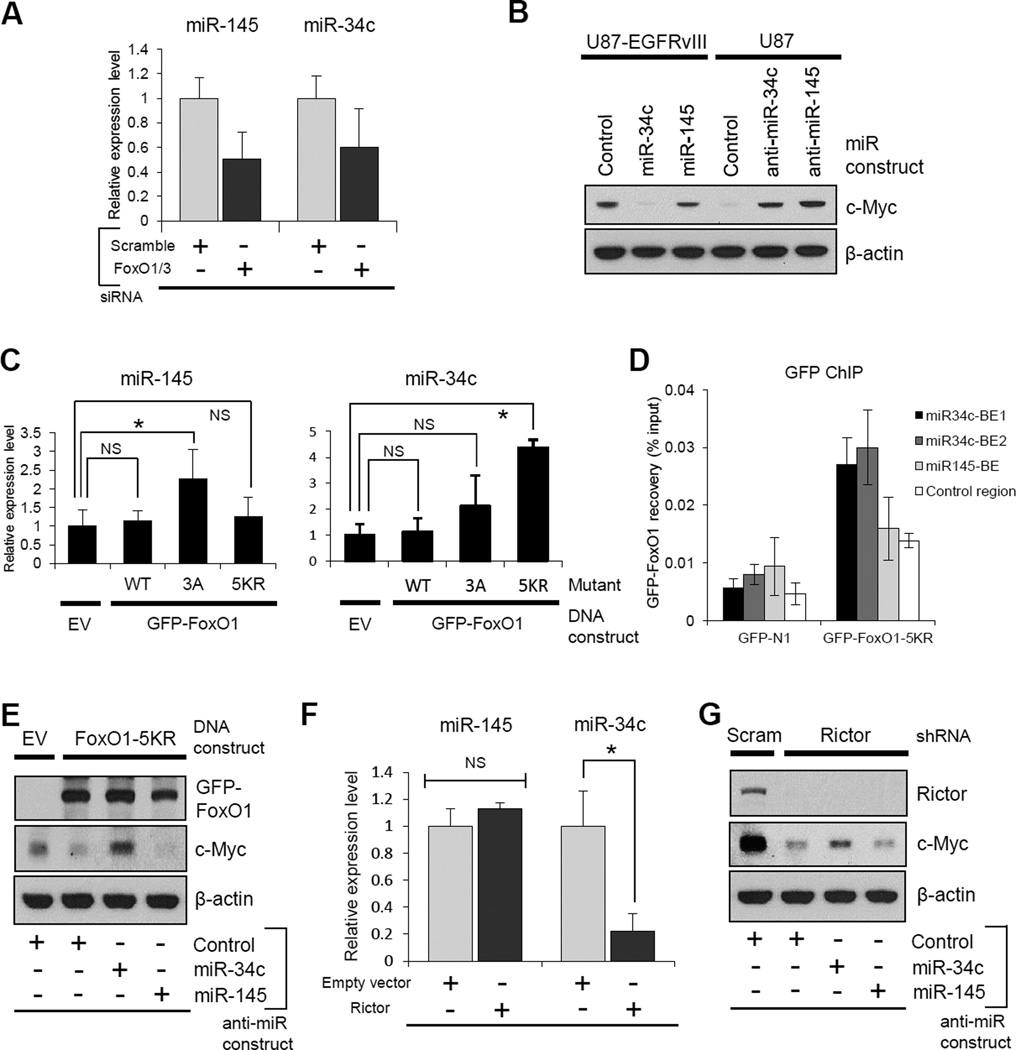
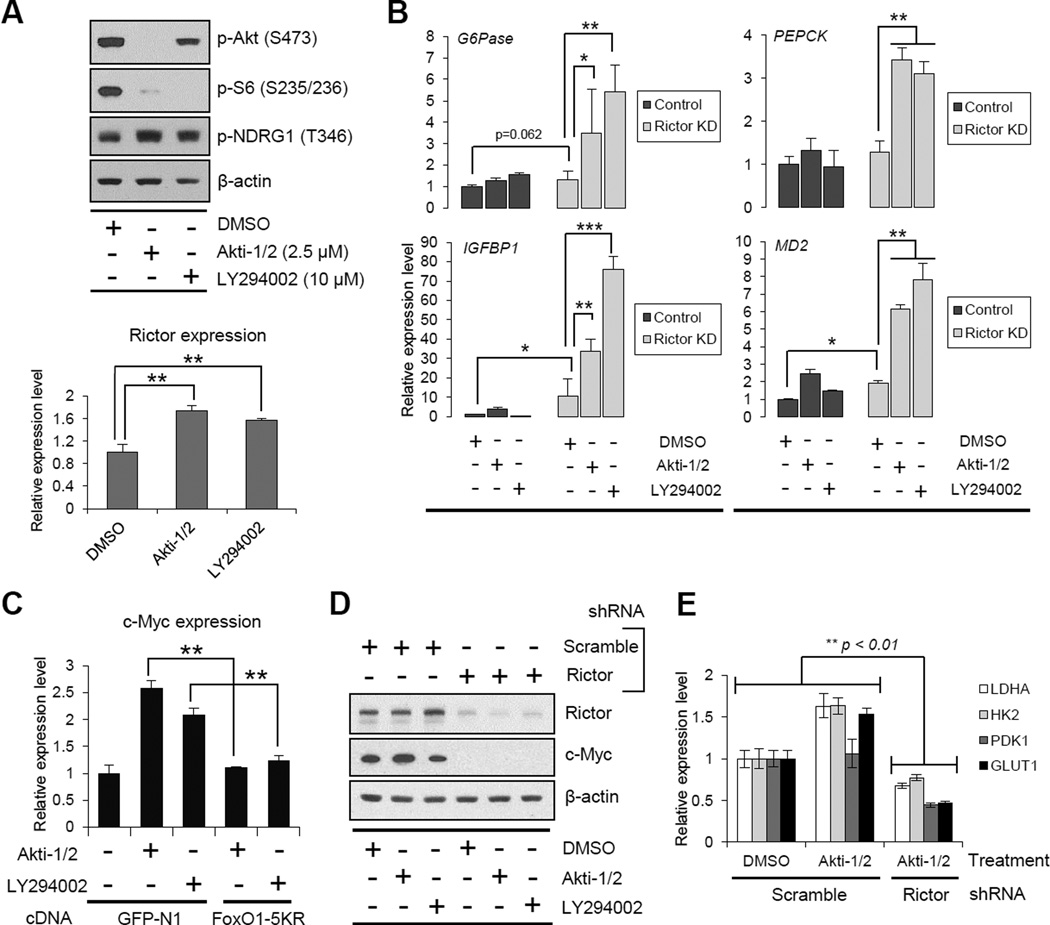
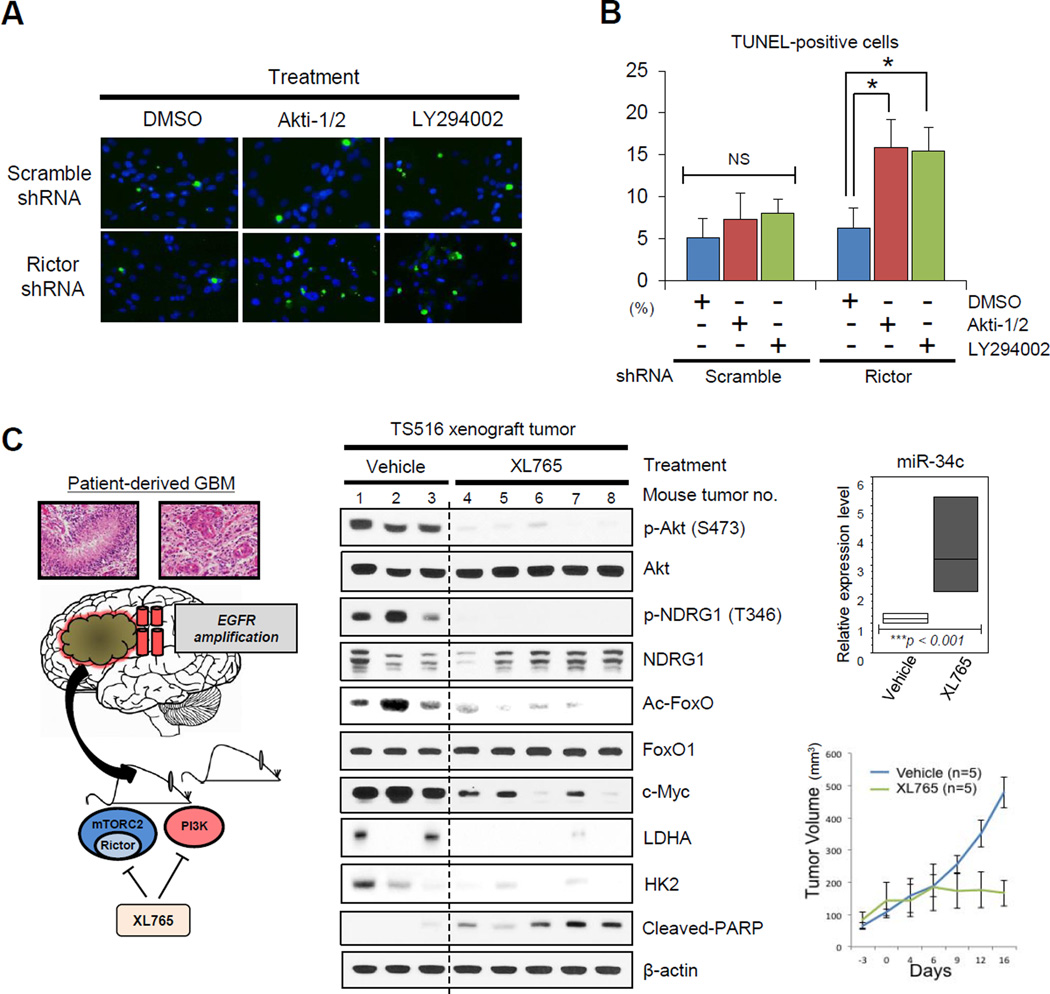
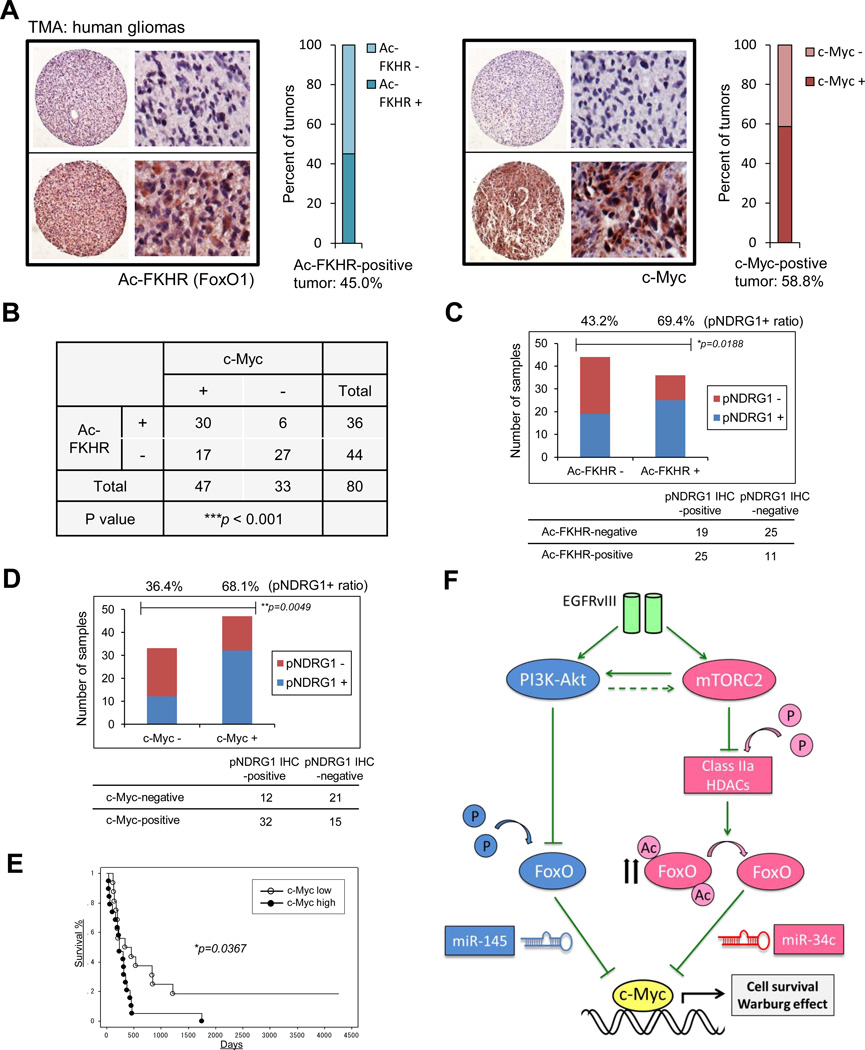
Aerobic glycolysis (the Warburg effect) is a core hallmark of cancer, but the molecular mechanisms underlying it remain unclear. Here, we identify an unexpected central role for mTORC2 in cancer metabolic reprogramming where it controls glycolytic metabolism by ultimately regulating the cellular level of c-Myc. We show that mTORC2 promotes inactivating phosphorylation of class IIa histone deacetylases, which leads to the acetylation of FoxO1 and FoxO3, and this in turn releases c-Myc from a suppressive miR-34c-dependent network. These central features of activated mTORC2 signaling, acetylated FoxO, and c-Myc levels are highly intercorrelated in clinical samples and with shorter survival of GBM patients. These results identify a specific, Akt-independent role for mTORC2 in regulating glycolytic metabolism in cancer.
Copyright © 2013 Elsevier Inc. All rights reserved.
Conflict of interest statement
All authors report no conflict of interest.
Figures
Comment in
-
mTORC2 dictates Warburg effect and drug resistance.Cell Cycle. 2014;13(7):1053-4. doi: 10.4161/cc.28377. Epub 2014 Feb 28. Cell Cycle. 2014. PMID: 24583874 Free PMC article. No abstract available.
References
-
- Albihn A, Johnsen JI, Henriksson MA. MYC in oncogenesis and as a target for cancer therapies. Adv Cancer Res. 2010;107:163–224. - PubMed
-
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. 2006. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
