

Reassessing target antigens for adoptive T-cell therapy
- PMID: 24142051
- PMCID: PMC4280065
- DOI: 10.1038/nbt.2725
Reassessing target antigens for adoptive T-cell therapy
Abstract
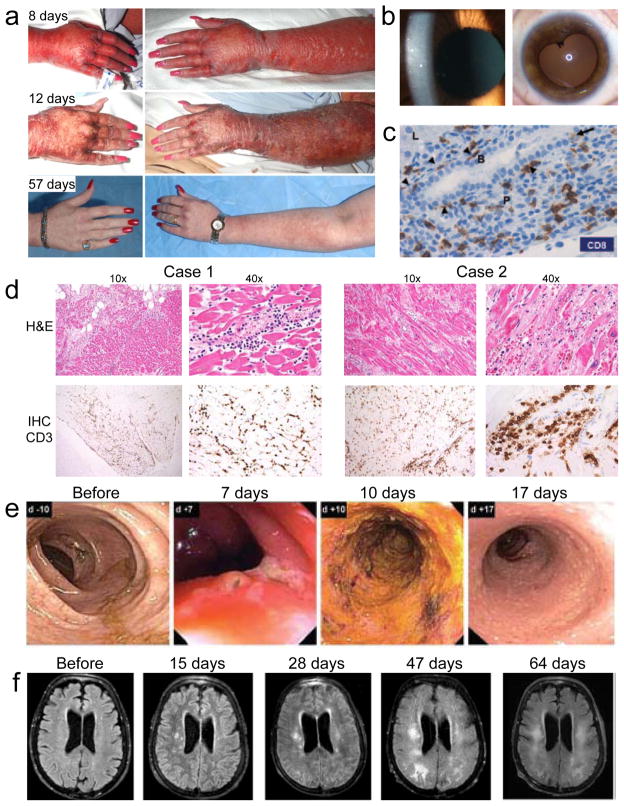
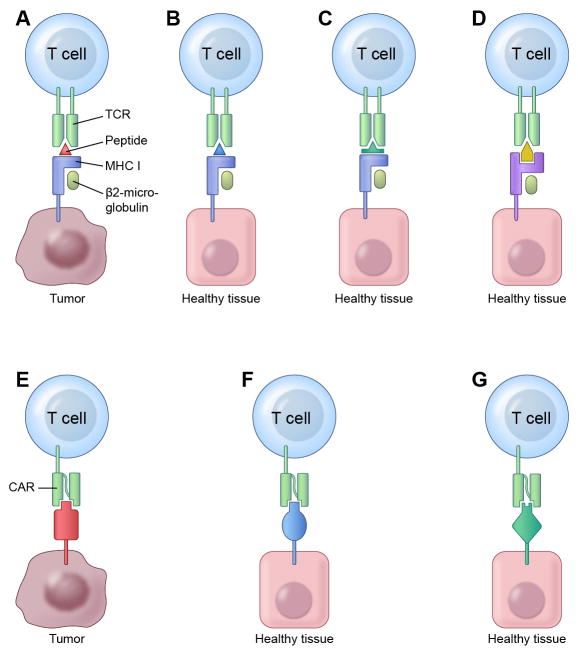
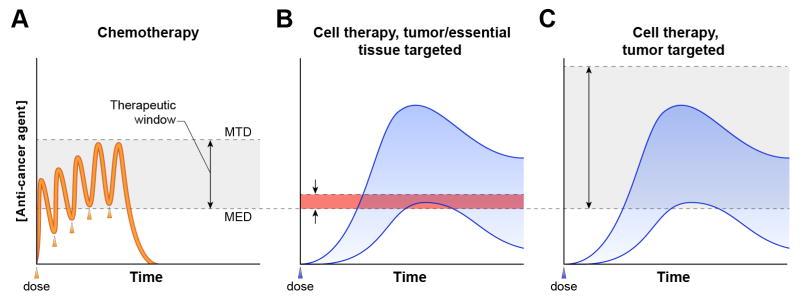
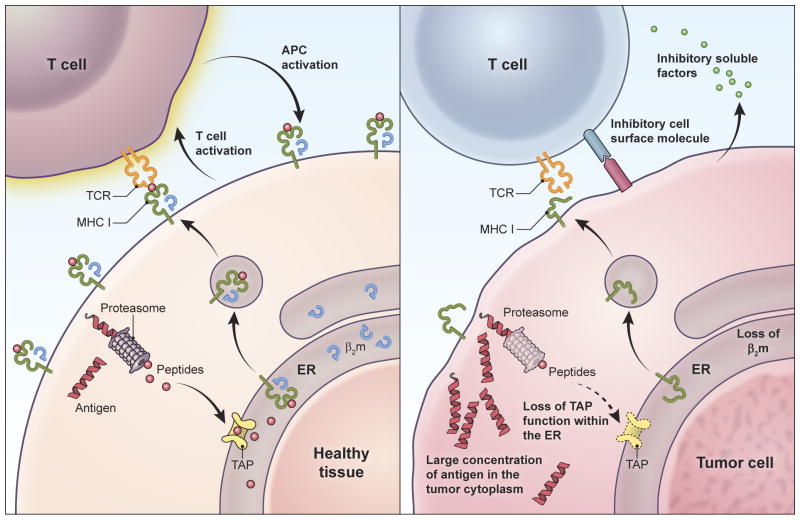
Adoptive T-cell therapy can target and kill widespread malignant cells thereby inducing durable clinical responses in melanoma and selected other malignances. However, many commonly targeted tumor antigens are also expressed by healthy tissues, and T cells do not distinguish between benign and malignant tissues if both express the target antigen. Autoimmune toxicity from T cell-mediated destruction of normal tissue has limited the development and adoption of this otherwise promising type of cancer therapy. A review of the unique biology of T-cell therapy and of recent clinical experience compels a reassessment of target antigens that traditionally have been viewed from the perspective of weaker immunotherapeutic modalities. It is important that target antigens chosen for adoptive T-cell therapy are expressed by tumors and not by essential healthy tissues. The risk of adverse autoimmune events can be further mitigated by generating antigen receptors using strategies that reduce the chance of cross-reactivity against epitopes in unintended targets. In general, a circumspect approach to target selection and thoughtful preclinical and clinical studies are pivotal to the ongoing advancement of these promising treatments.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
