

Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring
- PMID: 24142997
- PMCID: PMC3943309
- DOI: 10.3324/haematol.2013.093765
Next-generation sequencing-based multigene mutational screening for acute myeloid leukemia using MiSeq: applicability for diagnostics and disease monitoring
Abstract
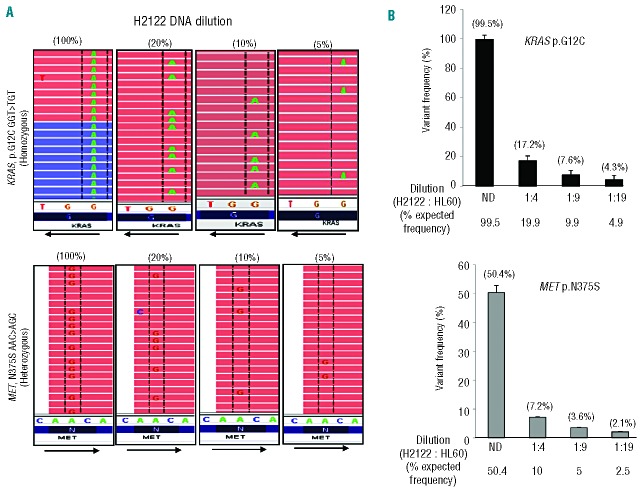
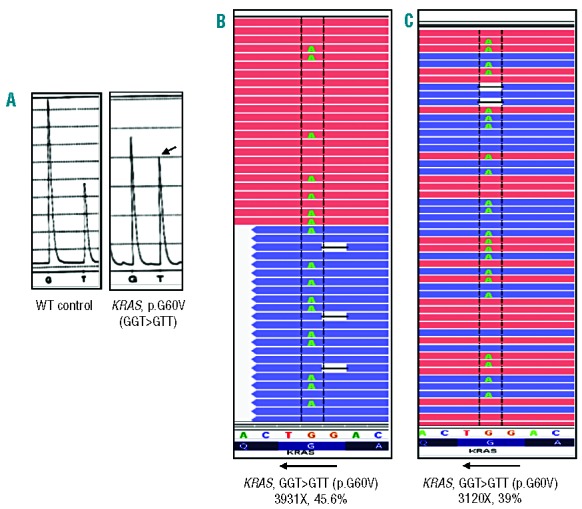
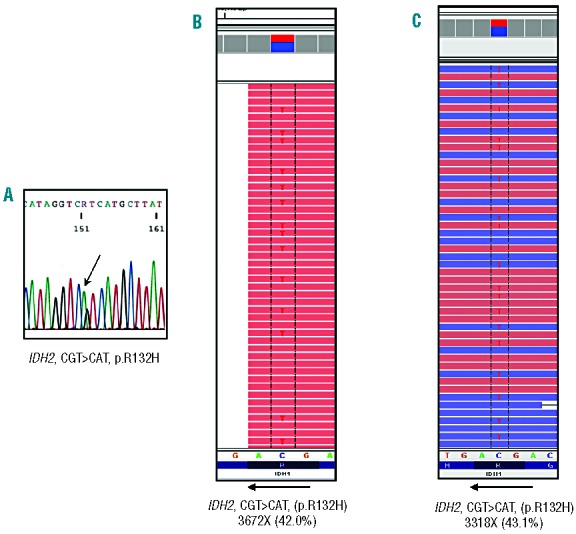
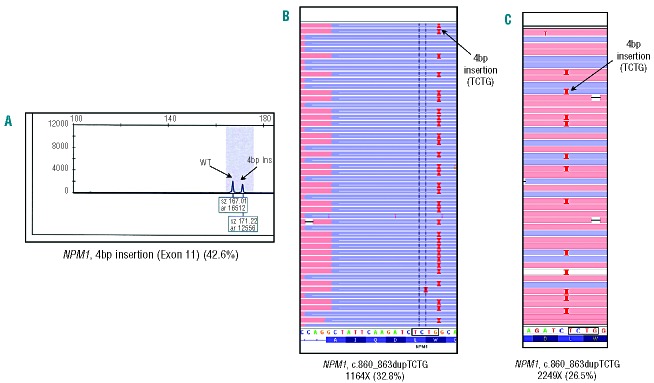
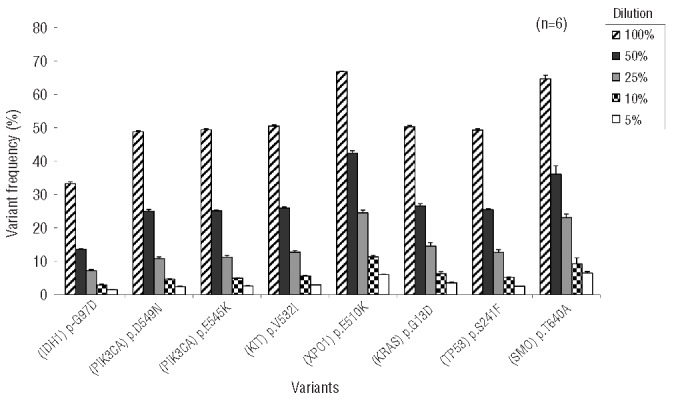
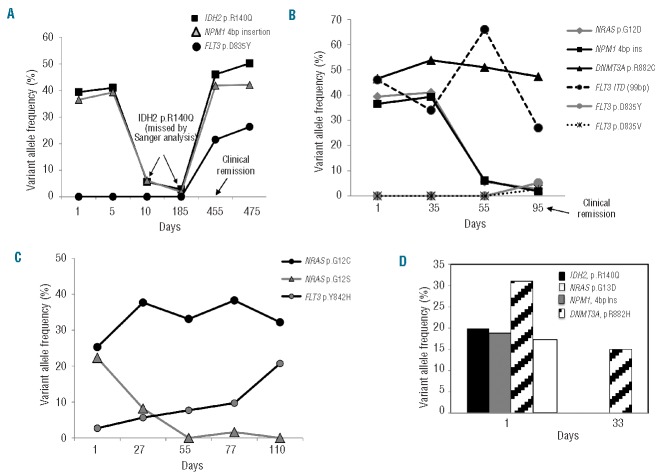
Routine molecular testing in acute myeloid leukemia involves screening several genes of therapeutic and prognostic significance for mutations. A comprehensive analysis using single-gene assays requires large amounts of DNA, is cumbersome and timely consolidation of results for clinical reporting is challenging. High throughput, next-generation sequencing platforms widely used in research have not been tested vigorously for clinical application. Here we describe the clinical application of MiSeq, a next-generation sequencing platform to screen mutational hotspots in 54 cancer-related genes including genes relevant in acute myeloid leukemia (NRAS, KRAS, FLT3, NPM1, DNMT3A, IDH1/2, JAK2, KIT and EZH2). We sequenced 63 samples from patients with acute myeloid leukemia/myelodysplastic syndrome using MiSeq and compared the results with those obtained using another next-generation sequencing platform, Ion-Torrent Personal Genome Machine and other conventional testing platforms. MiSeq detected a total of 100 single nucleotide variants and 23 NPM1 insertions that were confirmed by Ion Torrent or conventional platforms, indicating complete concordance. FLT3-internal tandem duplications (n=10) were not detected; however, re-analysis of the MiSeq output by Pindel, an indel detection algorithm, did detect them. Dilution studies of cancer cell-line DNA showed that the quantitative accuracy of mutation detection was up to an allelic frequency of 1.5% with a high level of inter- and intra-run assay reproducibility, suggesting potential utility for monitoring response to therapy, clonal heterogeneity and evolution. Examples demonstrating the advantages of MiSeq over conventional platforms for disease monitoring are provided. Easy work-flow, high throughput multiplexing capability, 4-day turnaround time and simultaneous assessment of routinely tested and emerging markers make MiSeq highly applicable for clinical molecular testing in acute myeloid leukemia.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
