

The one hour yeast proteome
- PMID: 24143002
- PMCID: PMC3879625
- DOI: 10.1074/mcp.M113.034769
The one hour yeast proteome
Abstract
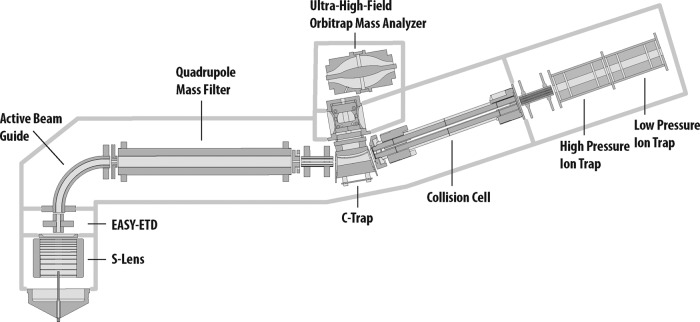
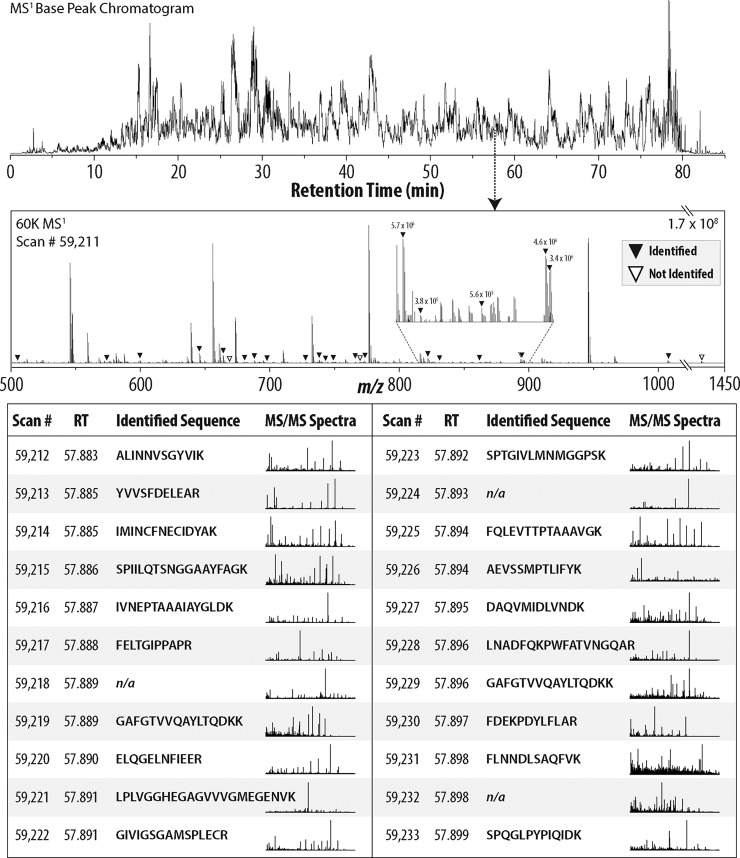
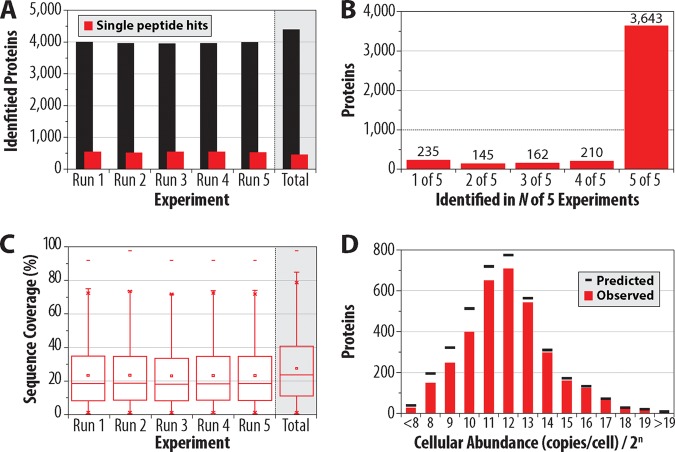
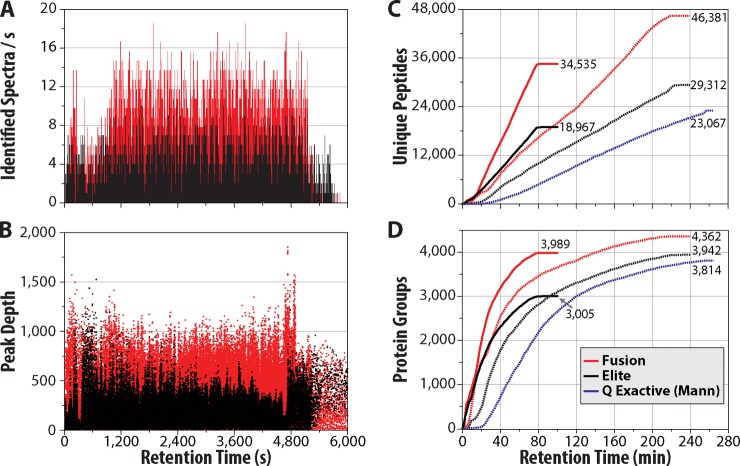
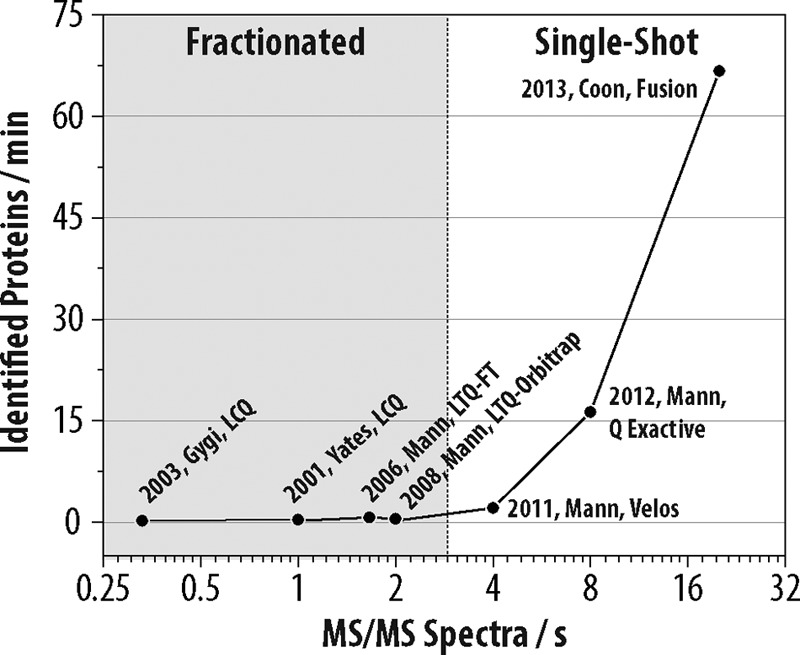
We describe the comprehensive analysis of the yeast proteome in just over one hour of optimized analysis. We achieve this expedited proteome characterization with improved sample preparation, chromatographic separations, and by using a new Orbitrap hybrid mass spectrometer equipped with a mass filter, a collision cell, a high-field Orbitrap analyzer, and, finally, a dual cell linear ion trap analyzer (Q-OT-qIT, Orbitrap Fusion). This system offers high MS(2) acquisition speed of 20 Hz and detects up to 19 peptide sequences within a single second of operation. Over a 1.3 h chromatographic method, the Q-OT-qIT hybrid collected an average of 13,447 MS(1) and 80,460 MS(2) scans (per run) to produce 43,400 (x) peptide spectral matches and 34,255 (x) peptides with unique amino acid sequences (1% false discovery rate (FDR)). On average, each one hour analysis achieved detection of 3,977 proteins (1% FDR). We conclude that further improvements in mass spectrometer scan rate could render comprehensive analysis of the human proteome within a few hours.
Figures
References
-
- Mallick P., Kuster B. (2010) Proteomics: a pragmatic perspective. Nat. Biotechnol. 28, 695–709 - PubMed
-
- Schena M., Shalon D., Davis R. W., Brown P. O. (1995) Quantitative monitoring of gene-expression patterns with a complementary-DNA microarray. Science 270, 467–470 - PubMed
-
- DeRisi J. L., Iyer V. R., Brown P. O. (1997) Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278, 680–686 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
