

Combining different mRNA capture methods to analyze the transcriptome: analysis of the Xenopus laevis transcriptome
- PMID: 24143257
- PMCID: PMC3797054
- DOI: 10.1371/journal.pone.0077700
Combining different mRNA capture methods to analyze the transcriptome: analysis of the Xenopus laevis transcriptome
Abstract
mRNA sequencing (mRNA-seq) is a commonly used technique to survey gene expression from organisms with fully sequenced genomes. Successful mRNA-seq requires purification of mRNA away from the much more abundant ribosomal RNA, which is typically accomplished by oligo-dT selection. However, mRNAs with short poly-A tails are captured poorly by oligo-dT based methods. We demonstrate that combining mRNA capture via oligo-dT with mRNA capture by the 5' 7-methyl guanosine cap provides a more complete view of the transcriptome and can be used to assay changes in mRNA poly-A tail length on a genome-wide scale. We also show that using mRNA-seq reads from both capture methods as input for de novo assemblers provides a more complete reconstruction of the transcriptome than either method used alone. We apply these methods of mRNA capture and de novo assembly to the transcriptome of Xenopus laevis, a well-studied frog that currently lacks a finished sequenced genome, to discover transcript sequences for thousands of mRNAs that are currently absent from public databases. The methods we describe here will be broadly applicable to many organisms and will provide insight into the transcriptomes of organisms with sequenced and unsequenced genomes.
Conflict of interest statement
Figures
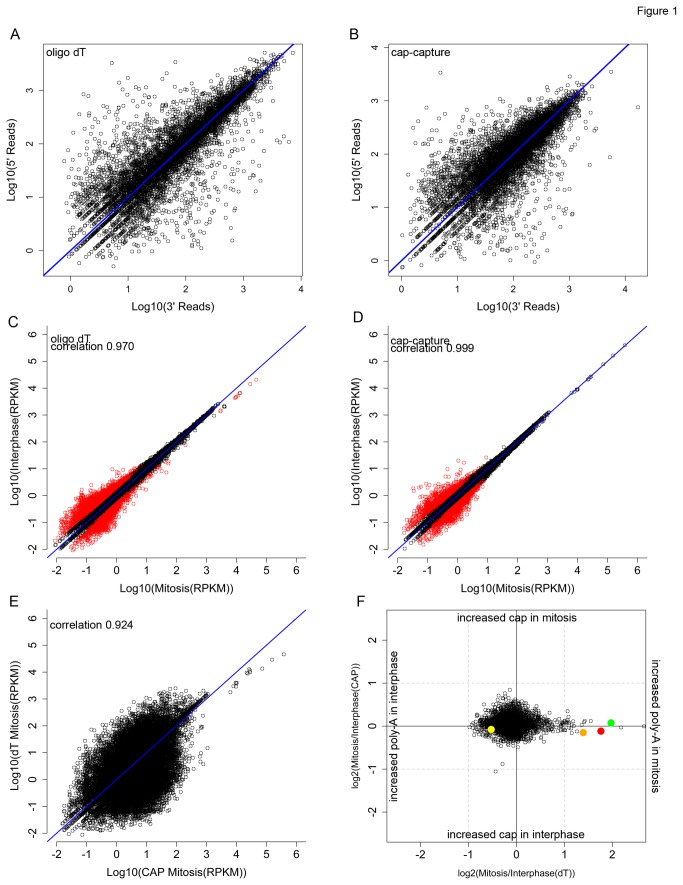
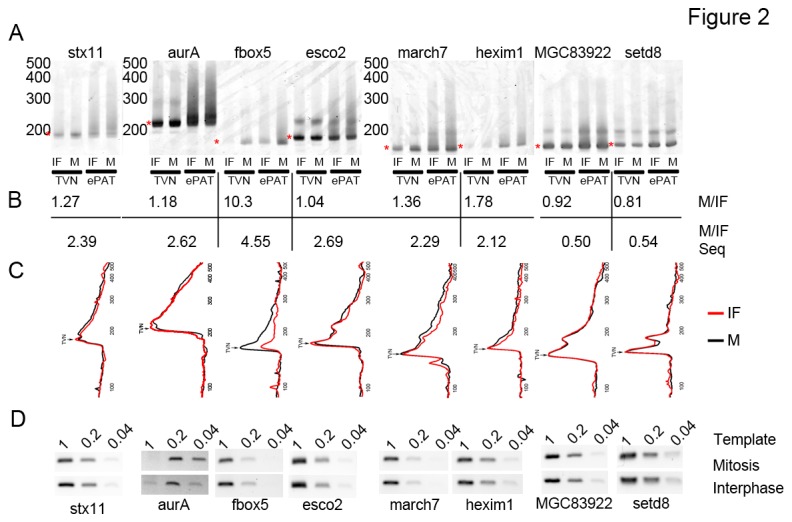
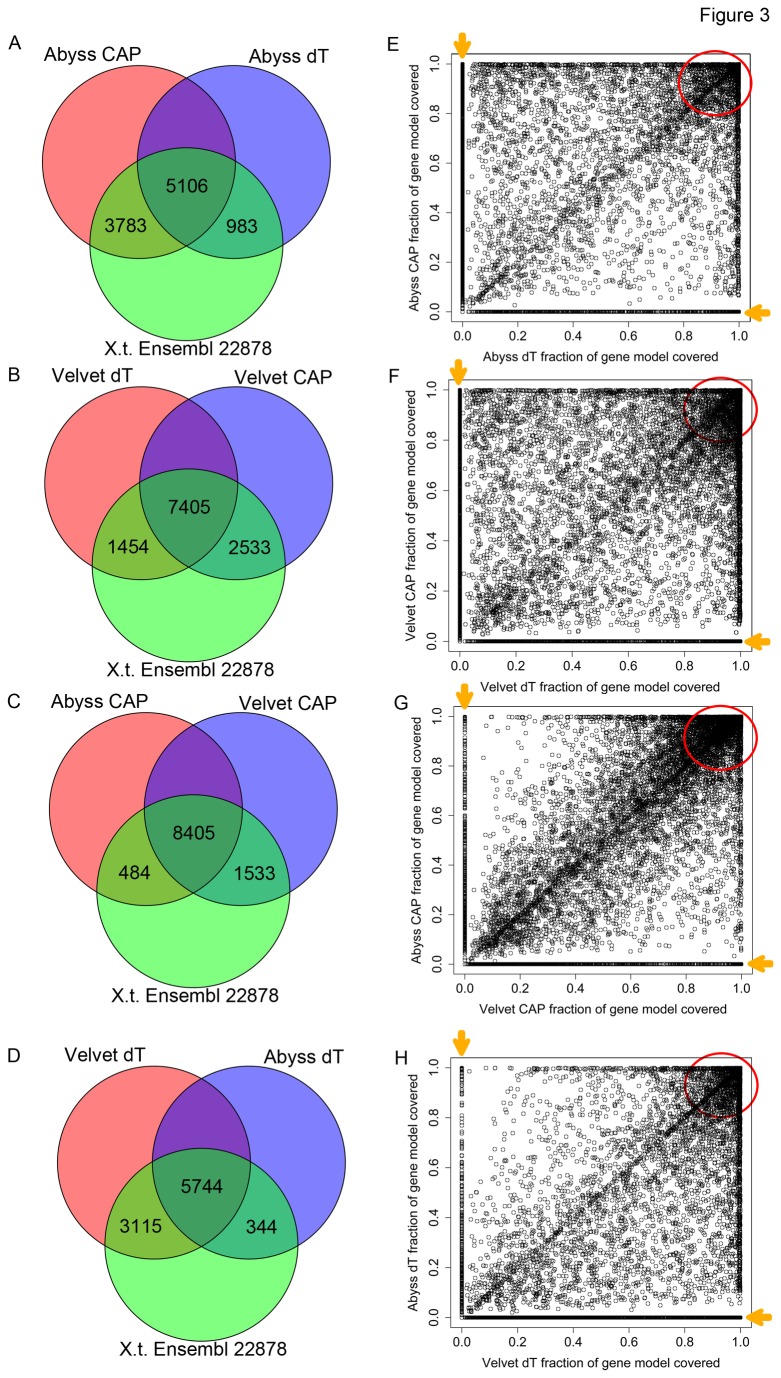
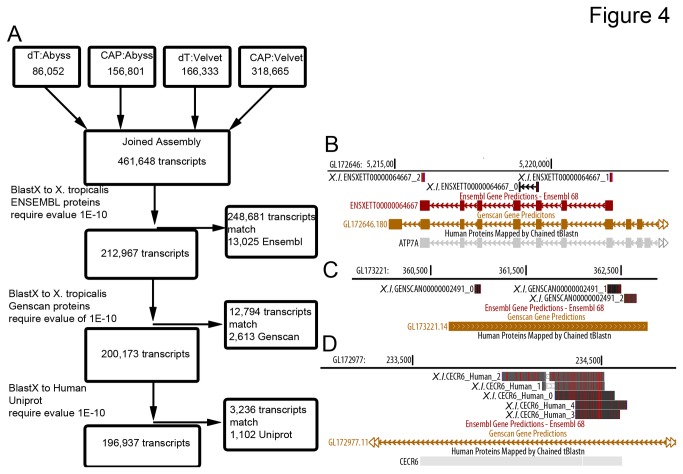
Similar articles
-
De novo Transcriptome Assemblies of Rana (Lithobates) catesbeiana and Xenopus laevis Tadpole Livers for Comparative Genomics without Reference Genomes.PLoS One. 2015 Jun 29;10(6):e0130720. doi: 10.1371/journal.pone.0130720. eCollection 2015. PLoS One. 2015. PMID: 26121473 Free PMC article.
-
Optimized design of antisense oligomers for targeted rRNA depletion.Nucleic Acids Res. 2021 Jan 11;49(1):e5. doi: 10.1093/nar/gkaa1072. Nucleic Acids Res. 2021. PMID: 33221877 Free PMC article.
-
A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing.BMC Genomics. 2017 May 22;18(1):395. doi: 10.1186/s12864-017-3757-8. BMC Genomics. 2017. PMID: 28532419 Free PMC article.
-
Current and Future Methods for mRNA Analysis: A Drive Toward Single Molecule Sequencing.Methods Mol Biol. 2018;1783:209-241. doi: 10.1007/978-1-4939-7834-2_11. Methods Mol Biol. 2018. PMID: 29767365 Review.
-
Transcriptome-wide measurement of poly(A) tail length and composition at subnanogram total RNA sensitivity by PAIso-seq.Nat Protoc. 2022 Sep;17(9):1980-2007. doi: 10.1038/s41596-022-00704-8. Epub 2022 Jul 13. Nat Protoc. 2022. PMID: 35831615 Review.
Cited by
-
Molecular asymmetry in the cephalochordate embryo revealed by single-blastomere transcriptome profiling.PLoS Genet. 2020 Dec 31;16(12):e1009294. doi: 10.1371/journal.pgen.1009294. eCollection 2020 Dec. PLoS Genet. 2020. PMID: 33382716 Free PMC article.
-
Genome-wide analysis identifies cis-acting elements regulating mRNA polyadenylation and translation during vertebrate oocyte maturation.RNA. 2020 Mar;26(3):324-344. doi: 10.1261/rna.073247.119. Epub 2020 Jan 2. RNA. 2020. PMID: 31896558 Free PMC article.
-
Combined ultra-low input mRNA and whole-genome sequencing of human embryonic stem cells.BMC Genomics. 2015 Nov 12;16:925. doi: 10.1186/s12864-015-2025-z. BMC Genomics. 2015. PMID: 26564201 Free PMC article.
-
Global approaches for profiling transcription initiation.Cell Rep Methods. 2021 Sep 27;1(5):100081. doi: 10.1016/j.crmeth.2021.100081. Epub 2021 Sep 16. Cell Rep Methods. 2021. PMID: 34632443 Free PMC article. Review.
-
An updated de novo transcriptome for green ash (Fraxinus pennsylvanica).G3 (Bethesda). 2023 Jun 1;13(6):jkad086. doi: 10.1093/g3journal/jkad086. G3 (Bethesda). 2023. PMID: 37070792 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
