

Prediction and experimental validation of enzyme substrate specificity in protein structures
- PMID: 24145433
- PMCID: PMC3831482
- DOI: 10.1073/pnas.1305162110
Prediction and experimental validation of enzyme substrate specificity in protein structures
Abstract
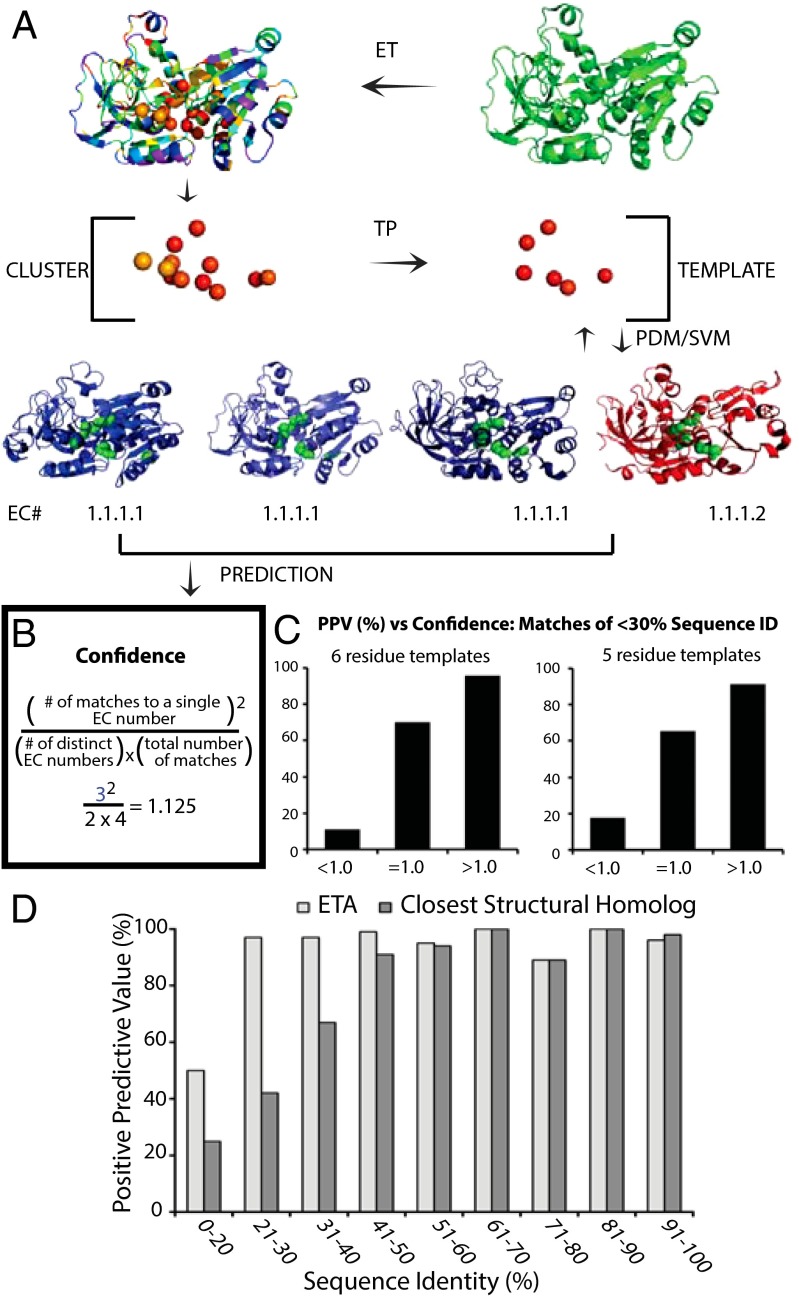
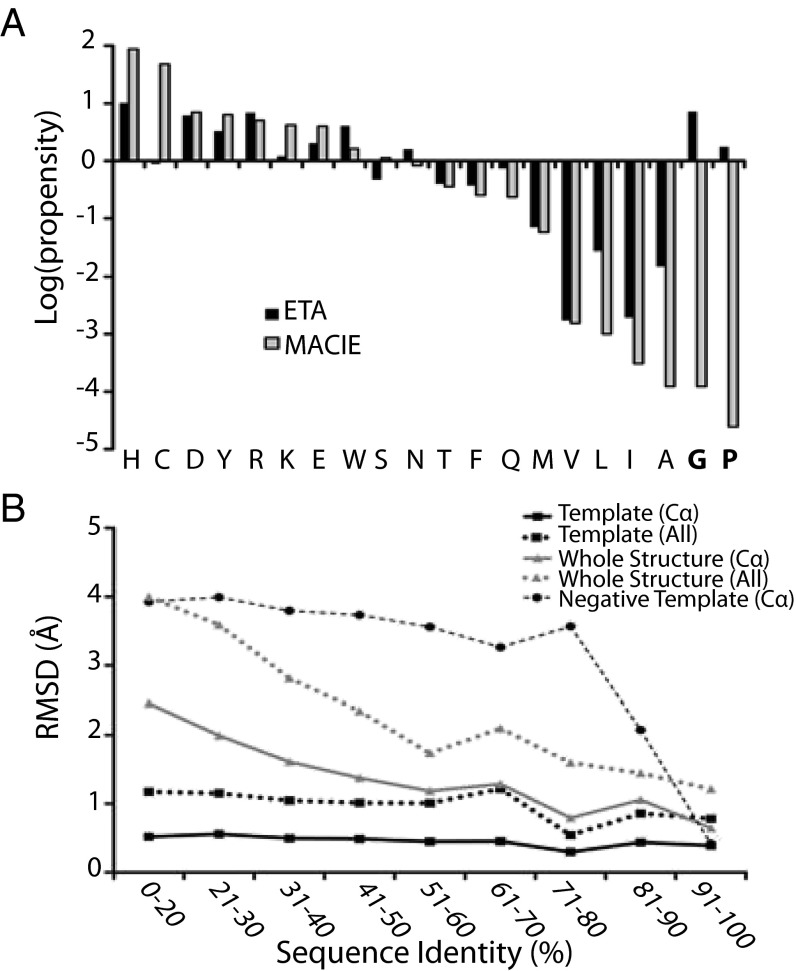
Structural Genomics aims to elucidate protein structures to identify their functions. Unfortunately, the variation of just a few residues can be enough to alter activity or binding specificity and limit the functional resolution of annotations based on sequence and structure; in enzymes, substrates are especially difficult to predict. Here, large-scale controls and direct experiments show that the local similarity of five or six residues selected because they are evolutionarily important and on the protein surface can suffice to identify an enzyme activity and substrate. A motif of five residues predicted that a previously uncharacterized Silicibacter sp. protein was a carboxylesterase for short fatty acyl chains, similar to hormone-sensitive-lipase-like proteins that share less than 20% sequence identity. Assays and directed mutations confirmed this activity and showed that the motif was essential for catalysis and substrate specificity. We conclude that evolutionary and structural information may be combined on a Structural Genomics scale to create motifs of mixed catalytic and noncatalytic residues that identify enzyme activity and substrate specificity.
Keywords: evolutionary trace; function annotation; protein function; structural motif.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
