

Review
doi: 10.1161/CIRCULATIONAHA.113.003199.
Targeting interleukin-1 in heart disease
Affiliations
- PMID: 24146121
- PMCID: PMC3938092
- DOI: 10.1161/CIRCULATIONAHA.113.003199
Item in Clipboard
Review
Targeting interleukin-1 in heart disease
Circulation.
.
No abstract available
Keywords: heart diseases; inflammation; interleukins.
Conflict of interest statement
Figures
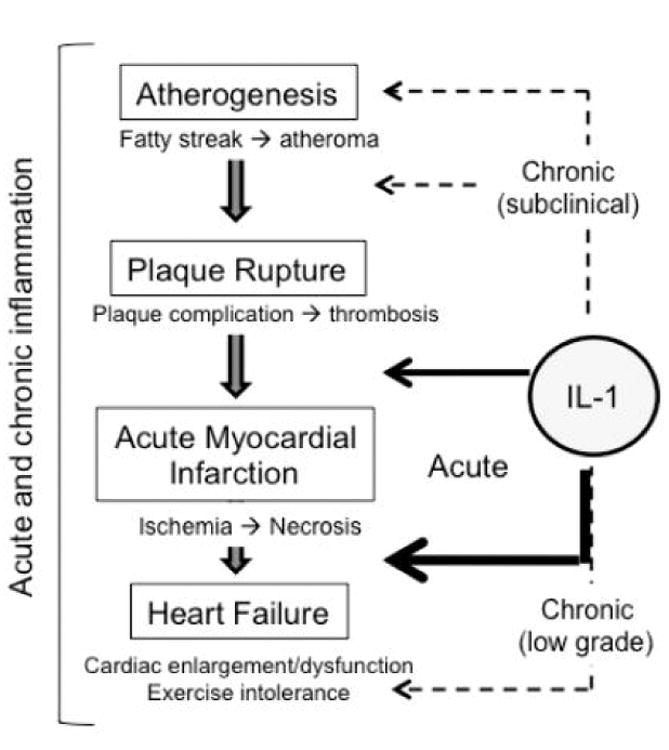
Inflammation and IL-1 in the wide spectrum of ischemic heart disease. The natural history of heart disease is not linear: evidence shows that atherogenesis is a slow process and a chronic subclinical inflammation may promote or accelerate the disease, whereas the atherothrombotic event leading to acute myocardial infarction is more of a sudden and rather stochastic event occurring on a predisposed substrate. An enhanced inflammatory response may predispose the plaques to rupture, and an even more intense response follows the myocardial injury affecting the degree of healing and the progression to heart failure. A chronic inflammation occurs also in many clinically stable patients with prior acute events and symptomatic for heart failure, potentially aggravating the cardiac dysfunction and/or predisposing to further decompensation.
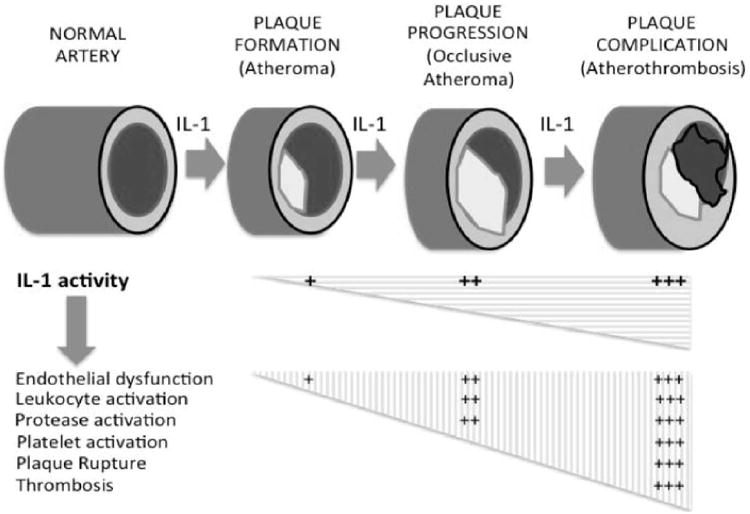
IL-1 and atherosclerosis. The natural history of atherosclerosis is characterized by plaque formation, progression (growth) and complication. IL-1 plays a key role in the formation, progression and complication of the atherosclerotic plaque. An increase in IL-1 activity causes a destabilization of the plaque, rupture and superimposed thrombus formation. Data excerpted from Abbate et al.
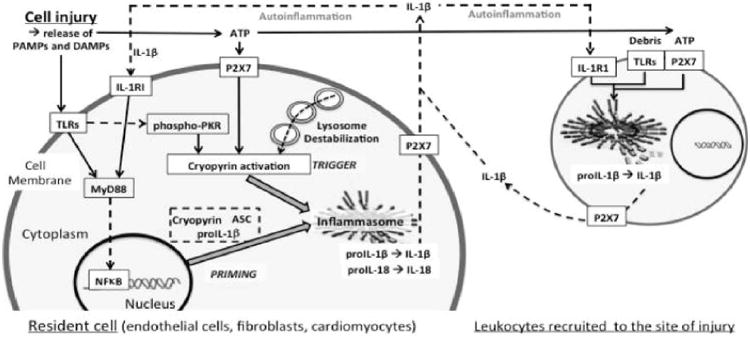
Cryopyrin activation and inflammasome formation. The figure shows a simplified scheme of cryopyrin activation and inflammasome formation in tissue resident cells and leukocytes recruited at the site of injury. TLR/IL-1RI agonists induce the inflammasome components (priming) through MyD88 signaling. ATP (through P2X7) or intracellular signals associated with lysosome destabilization activate cryopyrin triggering the formation of the inflammasome. The inflammasome processes and releases active IL-1β which further enhance the inflammatory response (autoinflammation). Data excerpted from Abbate et al.. Abbreviations: ASC=apoptosis-associated speck-like protein containing a C-terminal caspase-recruitment domain; ATP=adenosine triphosphate; DAMP= danger-associated molecular patterns; IL-1=Interleukin-1; IL-1RI=IL-1 receptor type I; MyD88=myeloid differentiation factor-88; NF-κB=nuclear factor-κB; PAMP=pathogen-associated molecular pattern; P2X7=purinergic receptor 2X7; PKR=RNA dependent protein kinase; TLR=Toll-like receptor.
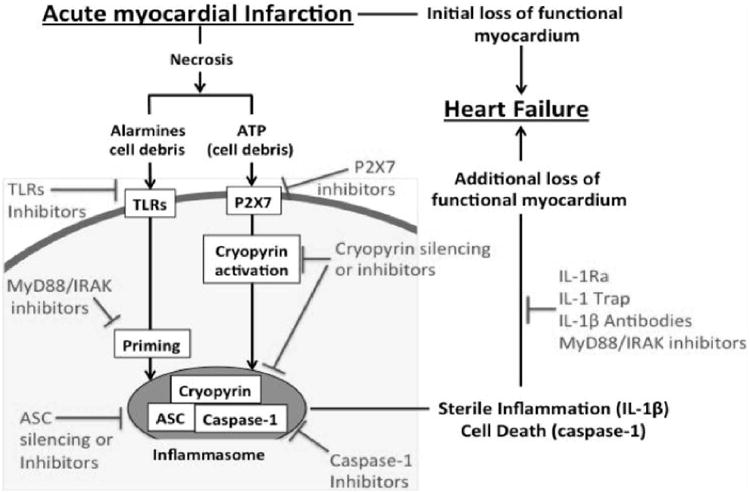
Inflammasome, IL-1 and heart failure following AMI. The formation of the cryopyrin inflammasome and subsequent caspase-1 activation promotes heart failure following acute myocardial infarction. The receptors which contribute to the inflammasome activation (TLR; P2X7; cryopyrin), the inflammasome components (cryopyrin, ASC and Caspase-1) and the inflammasome product IL-1β, and also contribute to the amplification of the inflammatory response and to cell death, are all potential targets for the treatment of acute myocardial infarction and the prevention or treatment of heart failure. Data excerpted from Mezzaroma et al.. Abbreviations: ASC=apoptosis-associated speck-like protein containing a C-terminal caspase-recruitment domain; AMI= acute myocardial infarction; ATP=adenosine triphosphate; IL-1=Interleukin-1; P2X7=purinergic receptor 2X7; TLR=Toll-like receptor; MyD88=myeloid differentiation factor-88; IRAK= interleukin-1 receptor associated kinase; IL-1Ra=Interleukin-1 receptor antagonist.
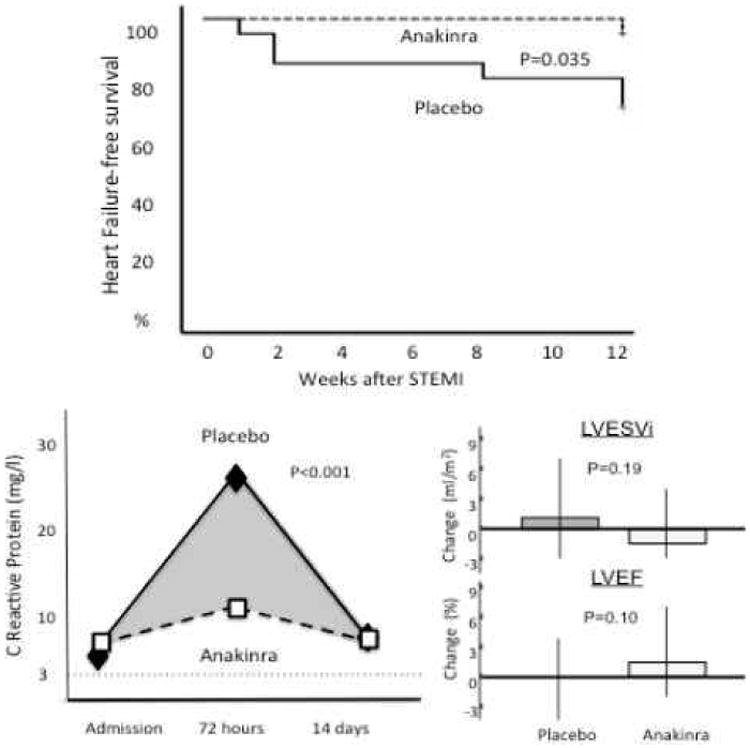
Interleukin-1 blockade in acute myocardial infarction. The VCU-ART and VCU-ART2 pilot studies showed significantly blunting of the inflammatory response with anakinra 100 mg daily during ST-segment elevation acute myocardial infarction and a numerically lower incidence of heart failure at 3 months. Data excerpted from Abbate et al.
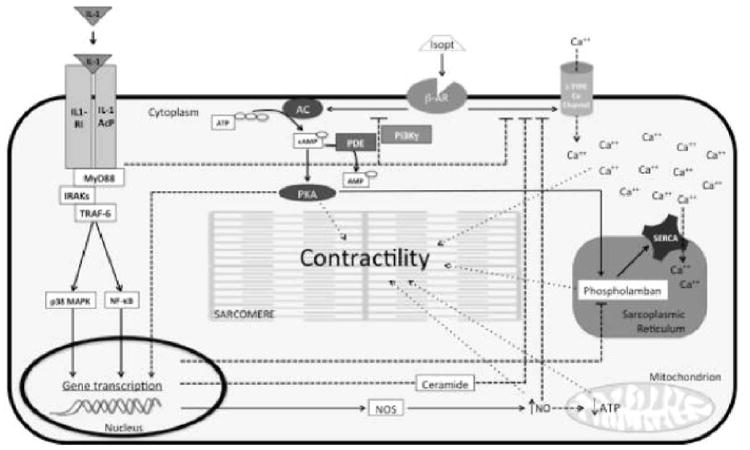
Proposed mechanisms of IL-1 induced contractile dysfunction. The diagram represents the intracellular pathways initiated by IL-1 and describes the proposed mechanisms by which IL-1 impairs the contractility of the cardiomyocyte. IL-1 (-α or -β) binds to the IL-1 receptor type I (IL-1RI) and the IL-1R accessory protein (IL-1AcP) and recruits the intracellular adaptor protein myeloid differentiation factor 88 (MyD88). This interaction activates the interleukin-1 receptor associated kinases (IRAKs) and the tumor necrosis factor (TNF) receptor associated factor 6 (TRAF-6) and leads to the activation of the p38 mitogen activated protein kinase (p38 MAPK) and the nuclear translocation of the nuclear factor kB (NF-kB), causing transcriptional changes of several genes. The IL-1 signaling induces uncoupling of the β-adrenergic receptor (β-AR) from the adenylyl cyclase (AC),, and from the L-type calcium (Ca++) channels,, thus reducing the cardiomyocytes responsiveness to the β-AR agonist (such as isoproterenol [isopt]). The cyclic AMP (cAMP)-dependent protein kinase A (PKA) is sensitive to increasing concentration of cAMP and regulates the function of several proteins, including sarcomere components, phospholamban and the calcium re-uptake in the sarcoplasmic reticulum, and the transcription of several genes. The changes in the gene transcription downstream of the IL-1 receptor affect the expression of phospholamban and the sarco/endoplasmic reticulum Ca++-ATPase (SERCA),, and increase the production of ceramide, which contributes to the uncoupling of the Ca++ channel from the β-AR. A similar effect has been described by increasing amounts of nitric oxide (NO) secondary to augmented expression of the nitric oxide synthase (NOS), - which not only contributes to the uncoupling of the Ca++ regulation from the β-AR, but also affects the production of ATP in the mitochondria. Altered levels of Ca++, cAMP (through PKA) and ATP affect the contractility of the sarcomeres, the cardiomyocytes and the heart as a whole. Class 3 cAMP phosphodiesterases (PDE-3) oppose the effects of AC by hydrolyzing cAMP into AMP. Phosphoinositide-3 kinase gamma (PI3Kγ) regulates the activity of β-AR and PDE-3. PI3Kγ is also activated downstream of the IL-1 receptor and may be involved in the impairment of contractility by activating PDE-3 , and inducing β-AR desensitization.
Comment in
-
Letter by Takahashi regarding article "targeting interleukin-1 in heart disease".Circulation. 2014 Aug 12;130(7):e62. doi: 10.1161/CIRCULATIONAHA.114.008658. Circulation. 2014. PMID: 25114192 No abstract available.
-
Response to letter regarding article, "targeting interleukin-1 in heart disease".Circulation. 2014 Aug 12;130(7):e63. doi: 10.1161/CIRCULATIONAHA.114.010274. Circulation. 2014. PMID: 25114193 No abstract available.
References
-
- Doria A, Zen M, Bettio S, Gatto M, Bassi N, Nalotto L, Ghirardello A, Iaccarino L, Punzi L. Autoinflammation and autoimmunity: bridging the divide. Autoimmun Rev. 2012;12:22–30. - PubMed
-
- Medzhitov R, Janeway CA., Jr Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
