

Super-resolution imaging of bacteria in a microfluidics device
- PMID: 24146850
- PMCID: PMC3797773
- DOI: 10.1371/journal.pone.0076268
Super-resolution imaging of bacteria in a microfluidics device
Abstract
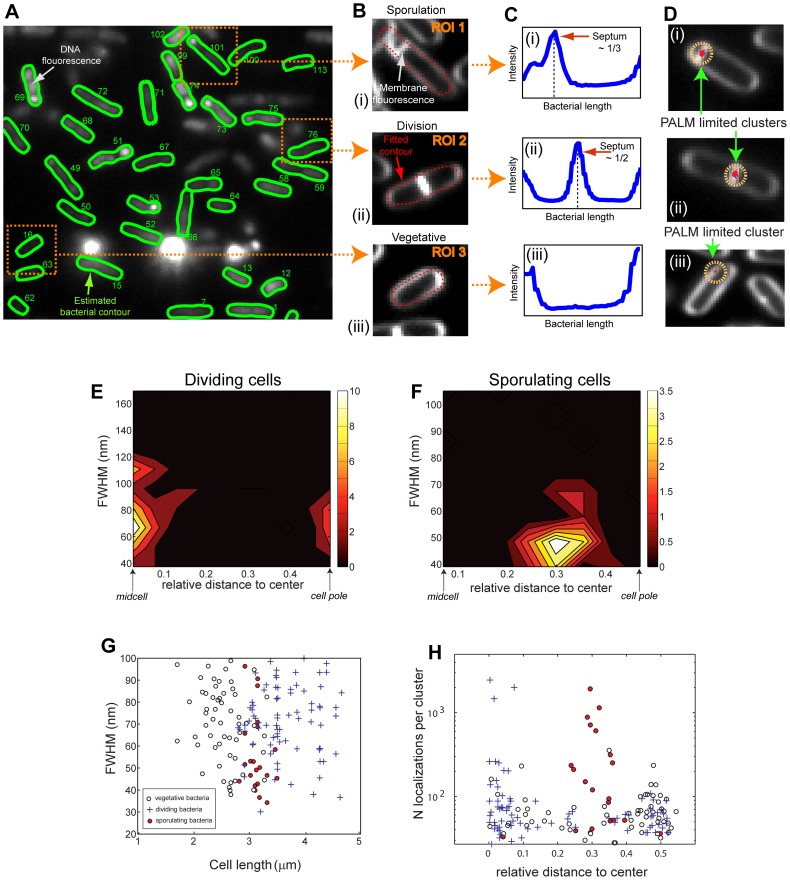
Bacteria have evolved complex, highly-coordinated, multi-component cellular engines to achieve high degrees of efficiency, accuracy, adaptability, and redundancy. Super-resolution fluorescence microscopy methods are ideally suited to investigate the internal composition, architecture, and dynamics of molecular machines and large cellular complexes. These techniques require the long-term stability of samples, high signal-to-noise-ratios, low chromatic aberrations and surface flatness, conditions difficult to meet with traditional immobilization methods. We present a method in which cells are functionalized to a microfluidics device and fluorophores are injected and imaged sequentially. This method has several advantages, as it permits the long-term immobilization of cells and proper correction of drift, avoids chromatic aberrations caused by the use of different filter sets, and allows for the flat immobilization of cells on the surface. In addition, we show that different surface chemistries can be used to image bacteria at different time-scales, and we introduce an automated cell detection and image analysis procedure that can be used to obtain cell-to-cell, single-molecule localization and dynamic heterogeneity as well as average properties at the super-resolution level.
Conflict of interest statement
Figures
Similar articles
-
Whole-cell multi-target single-molecule super-resolution imaging in 3D with microfluidics and a single-objective tilted light sheet.Nat Commun. 2024 Nov 24;15(1):10187. doi: 10.1038/s41467-024-54609-z. Nat Commun. 2024. PMID: 39582043 Free PMC article.
-
DNA Organization and Superesolved Segregation.Methods Mol Biol. 2018;1805:271-289. doi: 10.1007/978-1-4939-8556-2_14. Methods Mol Biol. 2018. PMID: 29971723
-
Three dimensional drift control at nano-scale in single molecule localization microscopy.Opt Express. 2020 Oct 26;28(22):32750-32763. doi: 10.1364/OE.404123. Opt Express. 2020. PMID: 33114953
-
Enhancing single molecule imaging in optofluidics and microfluidics.Int J Mol Sci. 2011;12(8):5135-56. doi: 10.3390/ijms12085135. Epub 2011 Aug 12. Int J Mol Sci. 2011. PMID: 21954349 Free PMC article. Review.
-
Eight years of single-molecule localization microscopy.Histochem Cell Biol. 2014 Jun;141(6):561-75. doi: 10.1007/s00418-014-1184-3. Epub 2014 Feb 5. Histochem Cell Biol. 2014. PMID: 24496595 Free PMC article. Review.
Cited by
-
RNA polymerase redistribution supports growth in E. coli strains with a minimal number of rRNA operons.Nucleic Acids Res. 2023 Aug 25;51(15):8085-8101. doi: 10.1093/nar/gkad511. Nucleic Acids Res. 2023. PMID: 37351576 Free PMC article.
-
Angular reconstitution-based 3D reconstructions of nanomolecular structures from superresolution light-microscopy images.Proc Natl Acad Sci U S A. 2017 Aug 29;114(35):9273-9278. doi: 10.1073/pnas.1704908114. Epub 2017 Aug 15. Proc Natl Acad Sci U S A. 2017. PMID: 28811371 Free PMC article.
-
Biochip with multi-planar electrodes geometry for differentiation of non-spherical bioparticles in a microchannel.Sci Rep. 2021 Jun 4;11(1):11880. doi: 10.1038/s41598-021-91109-2. Sci Rep. 2021. PMID: 34088942 Free PMC article.
-
Integrated multi-channel optical system for bacteria characterization and its potential use for monitoring of environmental bacteria.Biomed Opt Express. 2019 Feb 11;10(3):1165-1183. doi: 10.1364/BOE.10.001165. eCollection 2019 Mar 1. Biomed Opt Express. 2019. PMID: 30891337 Free PMC article.
-
Comparison of Escherichia coli surface attachment methods for single-cell microscopy.Sci Rep. 2019 Dec 19;9(1):19418. doi: 10.1038/s41598-019-55798-0. Sci Rep. 2019. PMID: 31857669 Free PMC article.
References
-
- Cozzarelli NR, Cost GJ, Nollmann M, Viard T, Stray JE (2006) Giant proteins that move DNA: bullies of the genomic playground. Nat Rev Mol Cell Biol 7: 580–588. - PubMed
-
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, et al. (2006) Imaging intracellular fluorescent proteins at nanometer resolution. Science 313: 1642–1645. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
