

Phylotranscriptomics: saturated third codon positions radically influence the estimation of trees based on next-gen data
- PMID: 24148944
- PMCID: PMC3845638
- DOI: 10.1093/gbe/evt157
Phylotranscriptomics: saturated third codon positions radically influence the estimation of trees based on next-gen data
Abstract
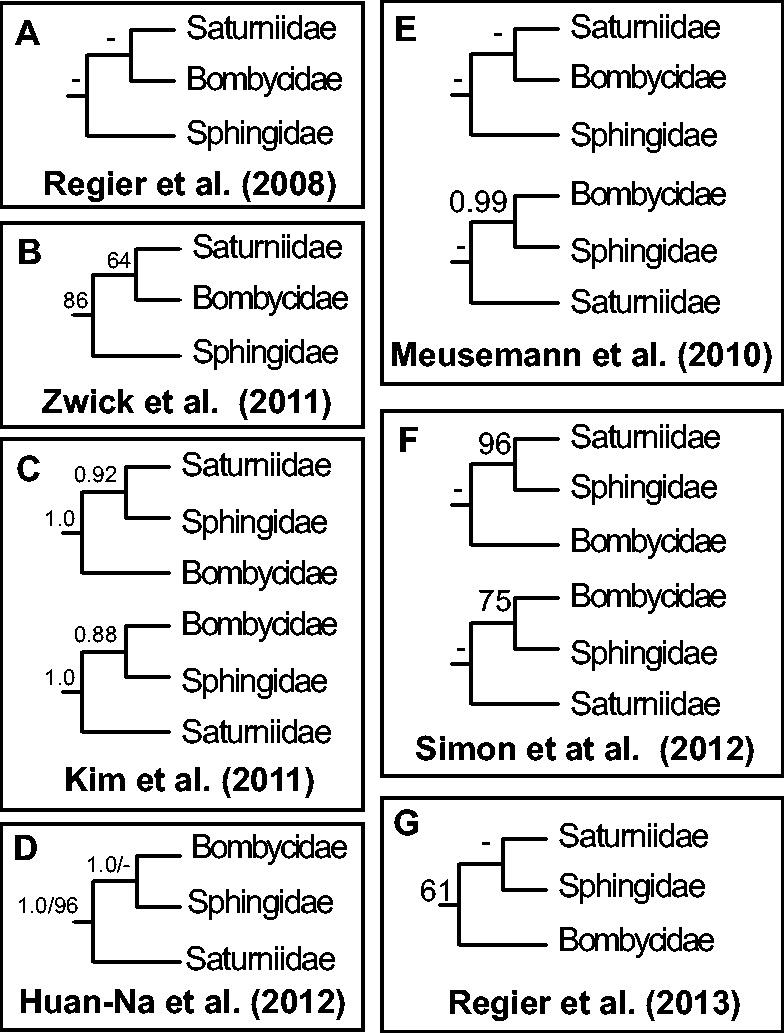
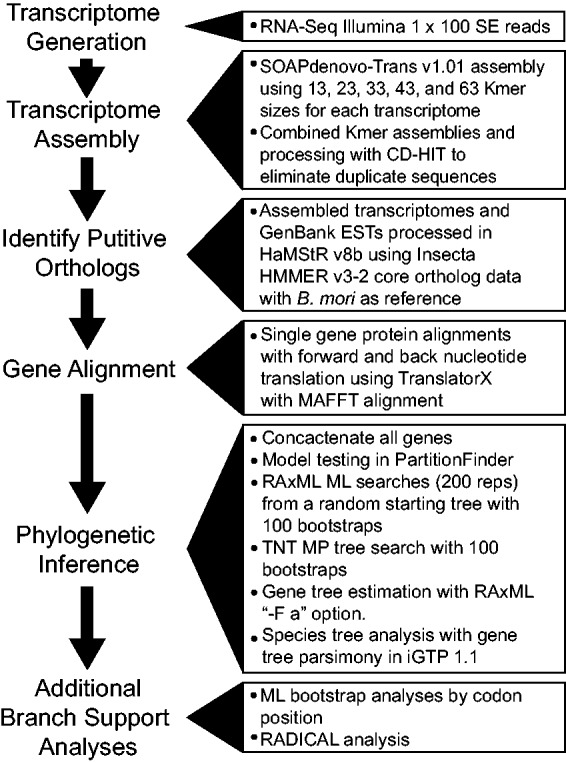
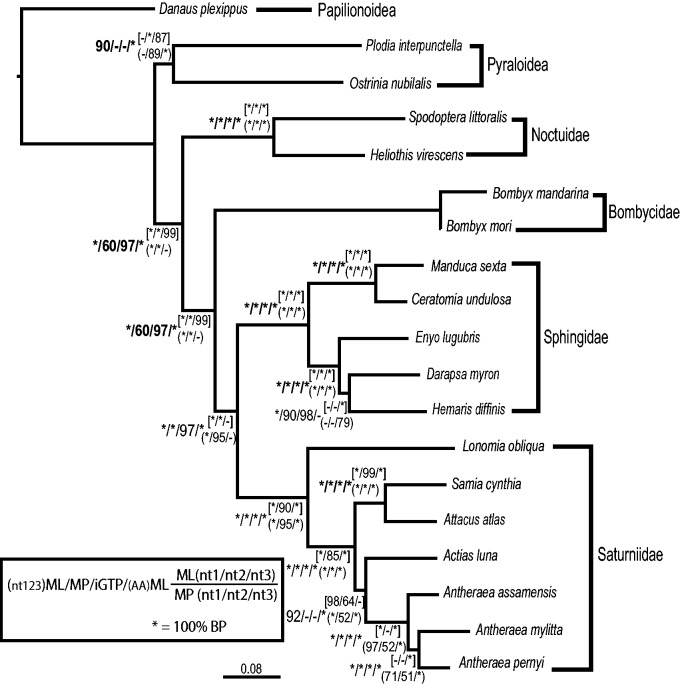
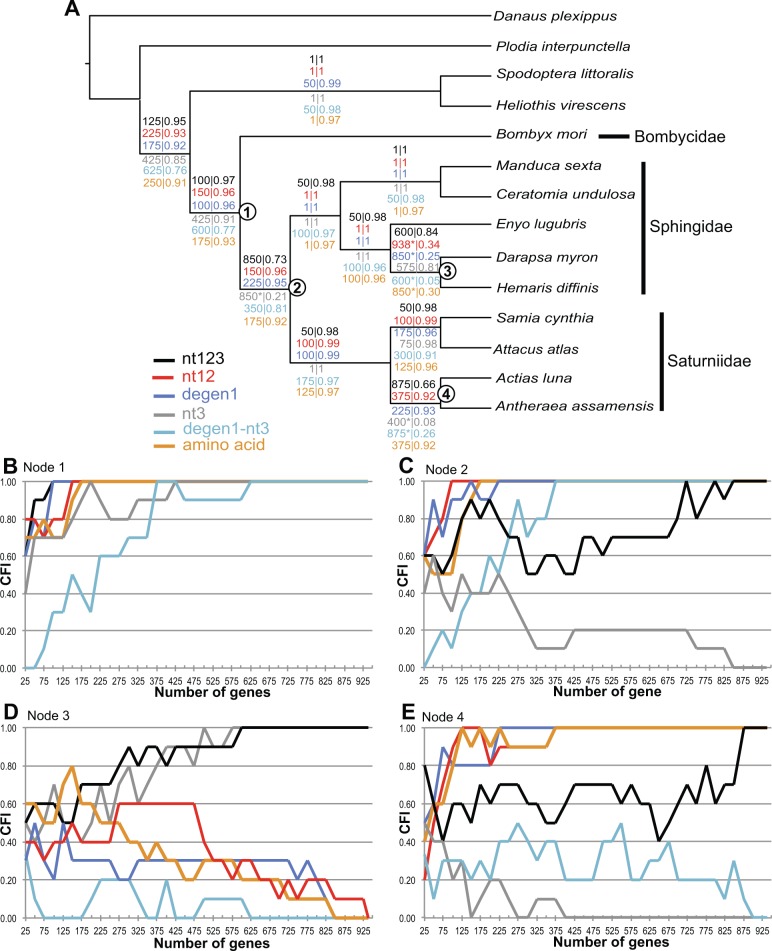
Recent advancements in molecular sequencing techniques have led to a surge in the number of phylogenetic studies that incorporate large amounts of genetic data. We test the assumption that analyzing large number of genes will lead to improvements in tree resolution and branch support using moths in the superfamily Bombycoidea, a group with some interfamilial relationships that have been difficult to resolve. Specifically, we use a next-gen data set that included 19 taxa and 938 genes (∼1.2M bp) to examine how codon position and saturation might influence resolution and node support among three key families. Maximum likelihood, parsimony, and species tree analysis using gene tree parsimony, on different nucleotide and amino acid data sets, resulted in largely congruent topologies with high bootstrap support compared with prior studies that included fewer loci. However, for a few shallow nodes, nucleotide and amino acid data provided high support for conflicting relationships. The third codon position was saturated and phylogenetic analysis of this position alone supported a completely different, potentially misleading sister group relationship. We used the program RADICAL to assess the number of genes needed to fix some of these difficult nodes. One such node originally needed a total of 850 genes but only required 250 when synonymous signal was removed. Our study shows that, in order to effectively use next-gen data to correctly resolve difficult phylogenetic relationships, it is necessary to assess the effects of synonymous substitutions and third codon positions.
Keywords: Bombycoidea; Lepidoptera; phylogeny; saturation; synonymous substitutions; transcriptome.
Figures
References
-
- Betancur-R R, Li C, Munroe TA, Ballesteros JA, Ortí G. Addressing gene tree discordance and non-stationarity to resolve a multi-locus phylogeny of the flatfishes (Teleostei: Pleuronectiformes) Syst Biol. 2013;62(5):763–785. - PubMed
-
- Biomatters. Geneious v5.5.8. 2013 [cited 2013 Nov 8]. Available from: http://www.geneious.com.
-
- Buckley TR, Simon C, Chambers GK. Exploring among-site rate variation models in a maximum likelihood framework using empirical data: the effects of model assumptions on estimates of topology, branch lengths, and bootstrap support. Syst Biol. 2001;50:67–86. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
