

Disclosing the crosstalk among DNA methylation, transcription factors, and histone marks in human pluripotent cells through discovery of DNA methylation motifs
- PMID: 24149073
- PMCID: PMC3847772
- DOI: 10.1101/gr.155960.113
Disclosing the crosstalk among DNA methylation, transcription factors, and histone marks in human pluripotent cells through discovery of DNA methylation motifs
Abstract
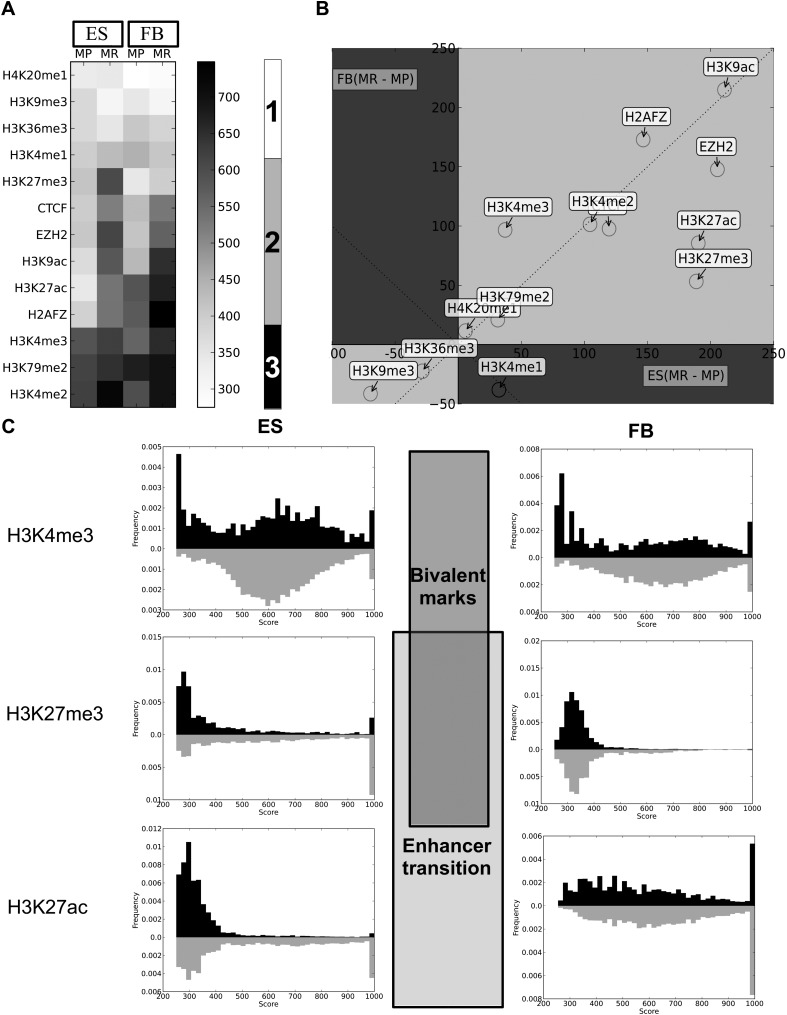
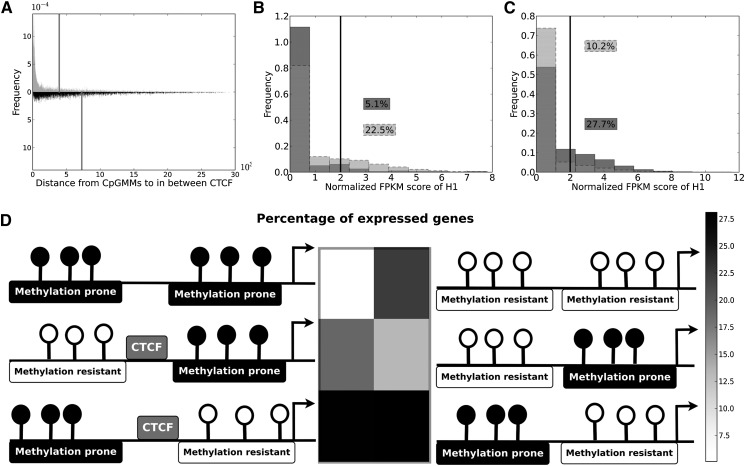
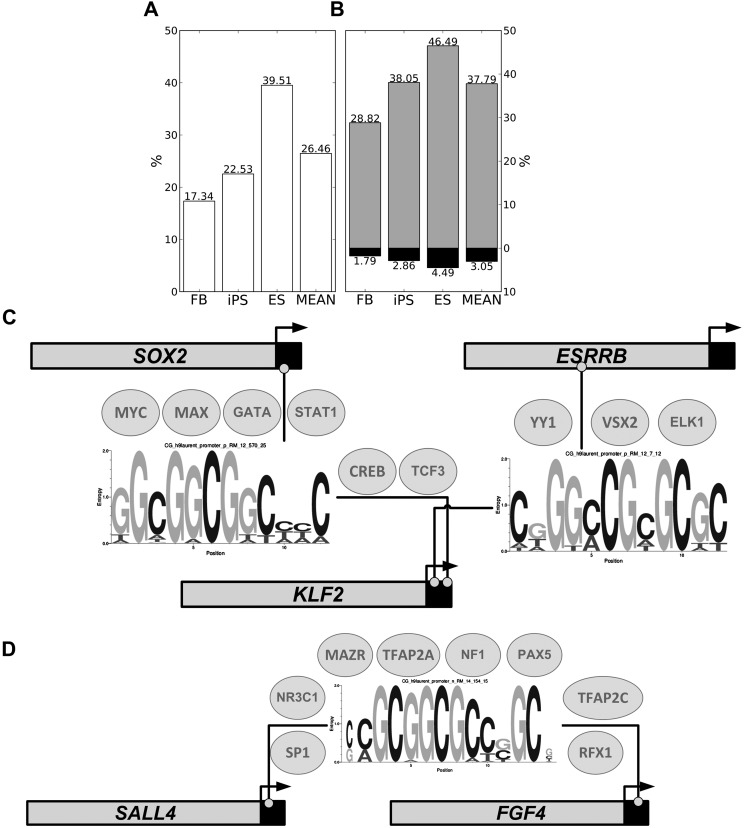
Gene expression regulation is gated by promoter methylation states modulating transcription factor binding. The known DNA methylation/unmethylation mechanisms are sequence unspecific, but different cells with the same genome have different methylomes. Thus, additional processes bringing specificity to the methylation/unmethylation mechanisms are required. Searching for such processes, we demonstrated that CpG methylation states are influenced by the sequence context surrounding the CpGs. We used such a property to develop a CpG methylation motif discovery algorithm. The newly discovered motifs reveal "methylation/unmethylation factors" that could recruit the "methylation/unmethylation machinery" to the loci specified by the motifs. Our methylation motif discovery algorithm provides a synergistic approach to the differently methylated region algorithms. Since our algorithm searches for commonly methylated regions inside the same sample, it requires only a single sample to operate. The motifs that were found discriminate between hypomethylated and hypermethylated regions. The hypomethylation-associated motifs have a high CG content, their targets appear in conserved regions near transcription start sites, they tend to co-occur within transcription factor binding sites, they are involved in breaking the H3K4me3/H3K27me3 bivalent balance, and they transit the enhancers from repressive H3K27me3 to active H3K27ac during ES cell differentiation. The new methylation motifs characterize the pluripotent state shared between ES and iPS cells. Additionally, we found a collection of motifs associated with the somatic memory inherited by the iPS from the initial fibroblast cells, thus revealing the existence of epigenetic somatic memory on a fine methylation scale.
Figures




References
-
- Berg OG, Von Hippel PH 1987. Selection of DNA binding sites by regulatory proteins: Statistical-mechanical theory and application to operators and promoters. J Mol Biol 193: 723–743 - PubMed
-
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326 - PubMed
-
- Bhasin M, Zhang H, Reinherz EL, Reche PA 2005. Prediction of methylated CpGs in DNA sequences using a support vector machine. FEBS Lett 579: 4302–4308 - PubMed
-
- Bird A 2011. Putting the DNA back into DNA methylation. Nat Genet 43: 1050–1051 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources