

Detecting range expansions from genetic data
- PMID: 24152007
- PMCID: PMC4282923
- DOI: 10.1111/evo.12202
Detecting range expansions from genetic data
Abstract
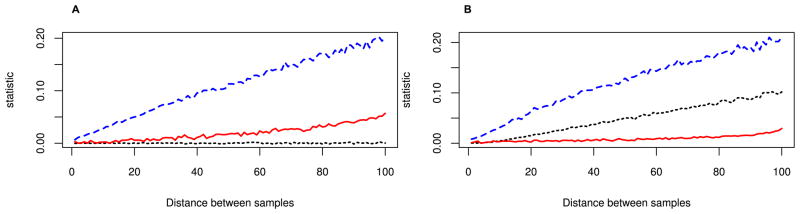
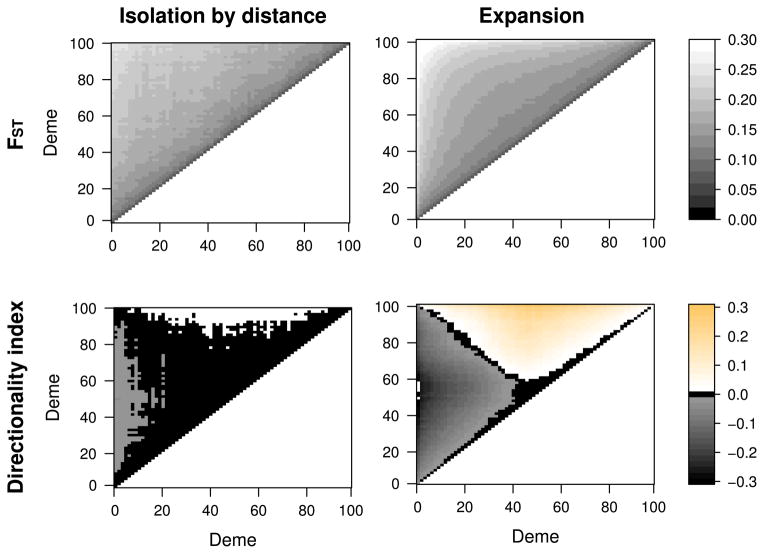
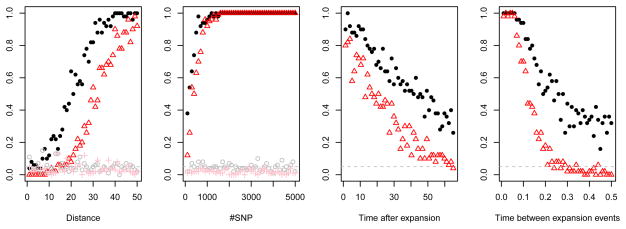
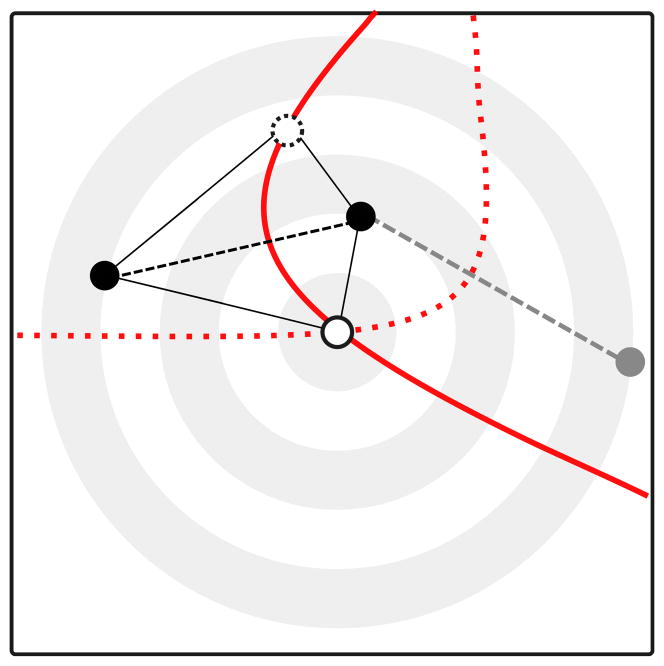
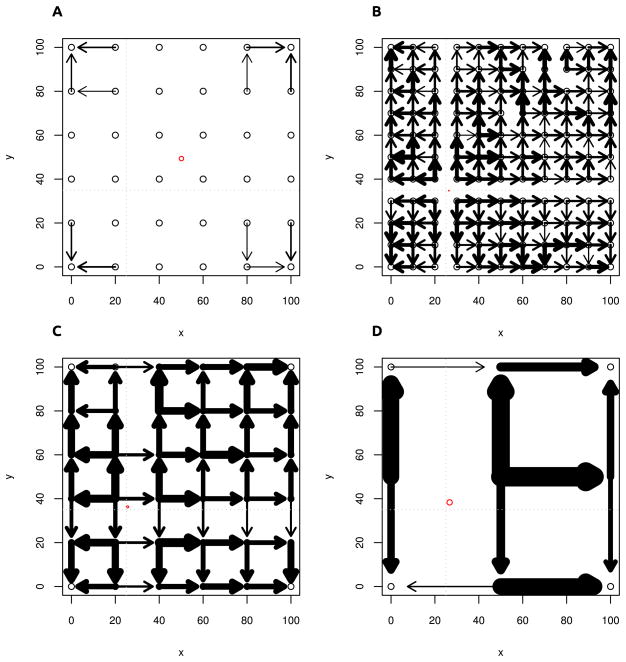
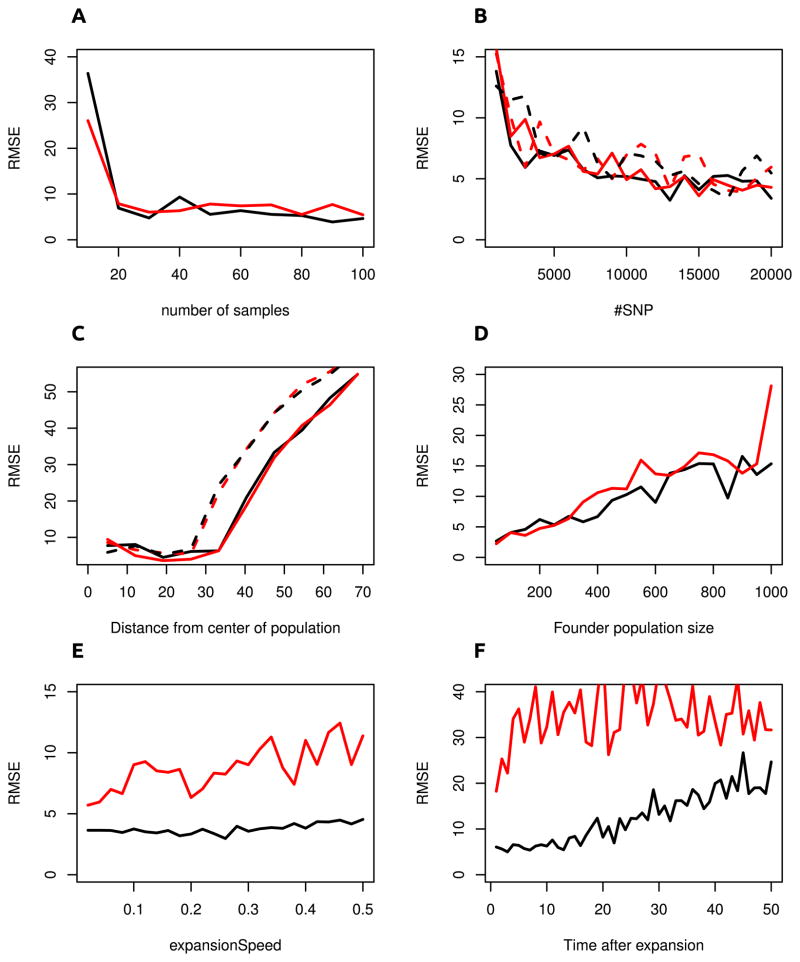
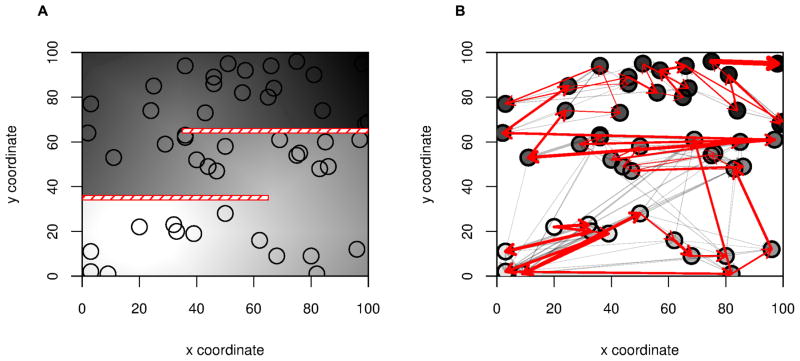
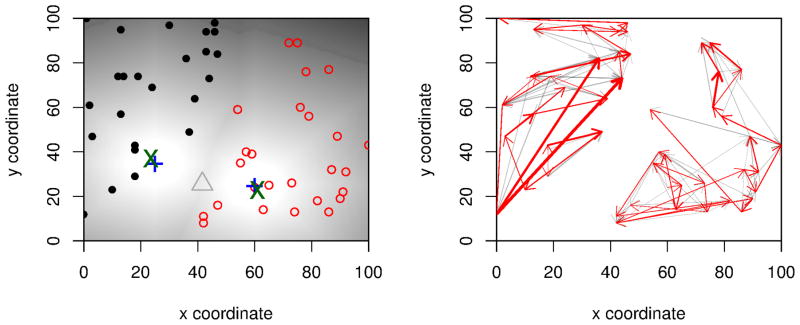
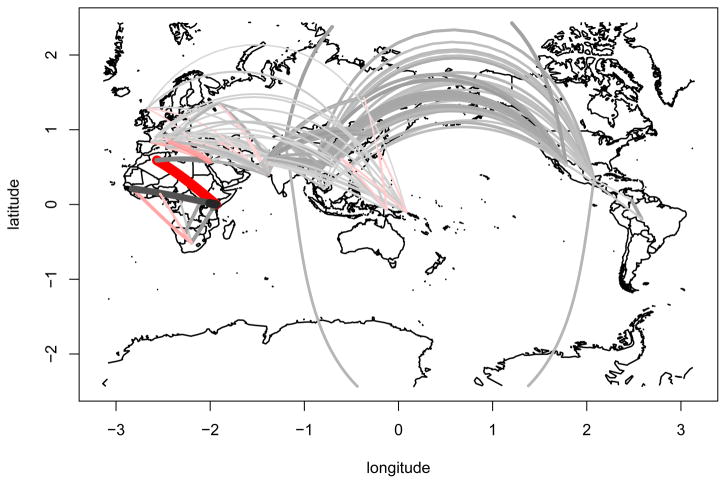
We propose a method that uses genetic data to test for the occurrence of a recent range expansion and to infer the location of the origin of the expansion. We introduce a statistic ψ (the directionality index) that detects asymmetries in the 2D allele frequency spectrum of pairs of population. These asymmetries are caused by the series of founder events that happen during an expansion and they arise because low frequency alleles tend to be lost during founder events, thus creating clines in the frequencies of surviving low-frequency alleles. Using simulations, we show that ψ is more powerful for detecting range expansions than both FST and clines in heterozygosity. We also show how we can adapt our approach to more complicated scenarios such as expansions with multiple origins or barriers to migration and we illustrate the utility of ψ by applying it to a data set from modern humans.
Keywords: Biogeography; evolutionary genomics; gene flow; genetic variation; population structure.
© 2013 The Author(s). Evolution © 2013 The Society for the Study of Evolution.
Figures
Similar articles
-
Boundary Effects Cause False Signals of Range Expansions in Population Genomic Data.Mol Biol Evol. 2024 May 3;41(5):msae091. doi: 10.1093/molbev/msae091. Mol Biol Evol. 2024. PMID: 38743590 Free PMC article.
-
The genetic signature of rapid range expansions: How dispersal, growth and invasion speed impact heterozygosity and allele surfing.Theor Popul Biol. 2014 Dec;98:1-10. doi: 10.1016/j.tpb.2014.08.005. Epub 2014 Sep 6. Theor Popul Biol. 2014. PMID: 25201435
-
Spatial genetic structure patterns of phenotype-limited and boundary-limited expanding populations: a simulation study.PLoS One. 2014 Jan 20;9(1):e85778. doi: 10.1371/journal.pone.0085778. eCollection 2014. PLoS One. 2014. PMID: 24465700 Free PMC article.
-
Nested clade analyses of phylogeographic data: testing hypotheses about gene flow and population history.Mol Ecol. 1998 Apr;7(4):381-97. doi: 10.1046/j.1365-294x.1998.00308.x. Mol Ecol. 1998. PMID: 9627999 Review.
-
Integrating the signatures of demic expansion and archaic introgression in studies of human population genomics.Curr Opin Genet Dev. 2016 Dec;41:140-149. doi: 10.1016/j.gde.2016.09.007. Epub 2016 Oct 13. Curr Opin Genet Dev. 2016. PMID: 27743539 Free PMC article. Review.
Cited by
-
Boundary Effects Cause False Signals of Range Expansions in Population Genomic Data.Mol Biol Evol. 2024 May 3;41(5):msae091. doi: 10.1093/molbev/msae091. Mol Biol Evol. 2024. PMID: 38743590 Free PMC article.
-
Range Expansion and the Origin of USA300 North American Epidemic Methicillin-Resistant Staphylococcus aureus.mBio. 2018 Jan 2;9(1):e02016-17. doi: 10.1128/mBio.02016-17. mBio. 2018. PMID: 29295910 Free PMC article.
-
Population genomic analyses support sympatric origins of parapatric morphs in a salamander.Ecol Evol. 2022 Nov 27;12(11):e9537. doi: 10.1002/ece3.9537. eCollection 2022 Nov. Ecol Evol. 2022. PMID: 36447598 Free PMC article.
-
Museum Skins Enable Identification of Introgression Associated with Cytonuclear Discordance.Syst Biol. 2024 Sep 5;73(3):579-593. doi: 10.1093/sysbio/syae016. Syst Biol. 2024. PMID: 38577768 Free PMC article.
-
Genetic architecture and evolution of color variation in American black bears.Curr Biol. 2023 Jan 9;33(1):86-97.e10. doi: 10.1016/j.cub.2022.11.042. Epub 2022 Dec 16. Curr Biol. 2023. PMID: 36528024 Free PMC article.
References
-
- Aho AV, Garey MR, Ullman JD. The Transitive Reduction of a Directed Graph. SIAM Journal on Computing. 1972;1:131–137. URL http://epubs.siam.org/doi/abs/10.1137/0201008. - DOI
-
- Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, Bonnen PE, De Bakker PI, Deloukas P, Gabriel SB. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52. URL http://europepmc.org/articles/PMC3173859. - PMC - PubMed
-
- Austerlitz F, Jung-Muller B, Godelle B, Gouyon PH. Evolution of coalescence times, genetic diversity and structure during colonization. Theoretical Population Biology. 1997;51:148–164.
-
- Balakrishnan V, Sanghvi LD. Distance between Populations on the Basis of Attribute Data. Biometrics. 1968;24:859–865. URL http://www.jstor.org/stable/2528876. ArticleType: research-article/Full publication date: Dec., 1968/Copyright © 1968 International Biometric Society.
-
- Beaumont MA, Zhang W, Balding DJ. Approximate Bayesian computation in population genetics. Genetics. 2002;162:2025–2035. URL http://www.ncbi.nlm.nih.gov/pubmed/12524368. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous