

DNA methylation is associated with an increased level of conservation at nondegenerate nucleotides in mammals
- PMID: 24157417
- PMCID: PMC3907051
- DOI: 10.1093/molbev/mst208
DNA methylation is associated with an increased level of conservation at nondegenerate nucleotides in mammals
Abstract
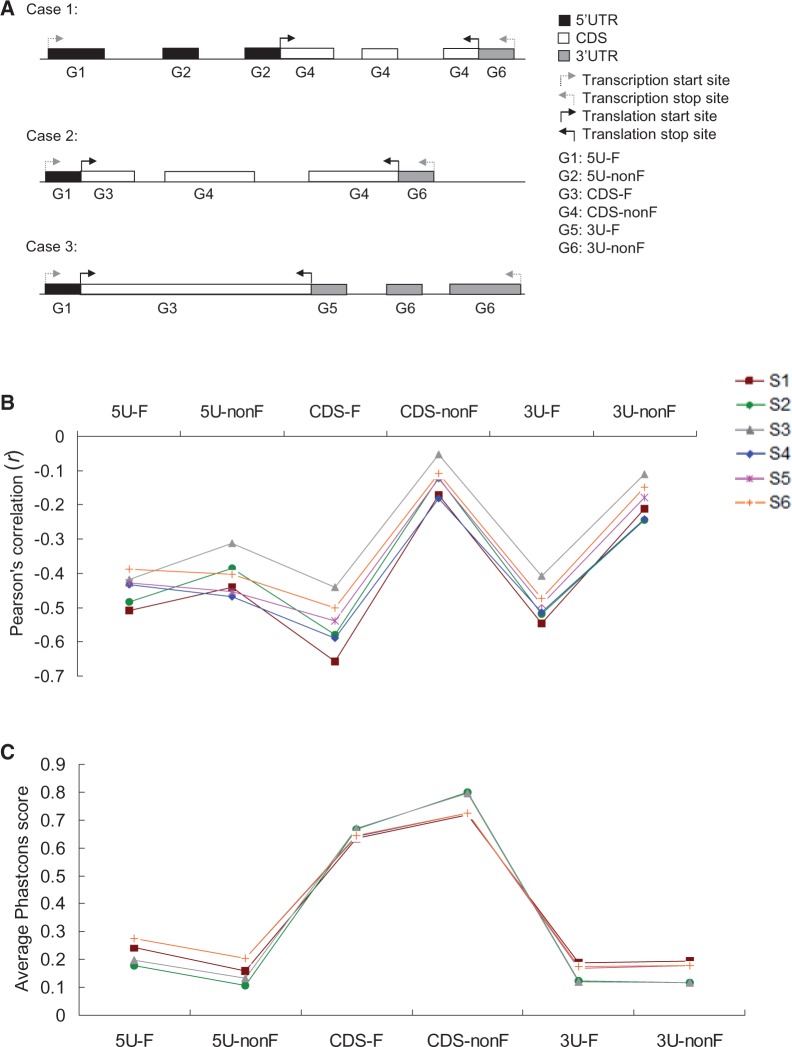
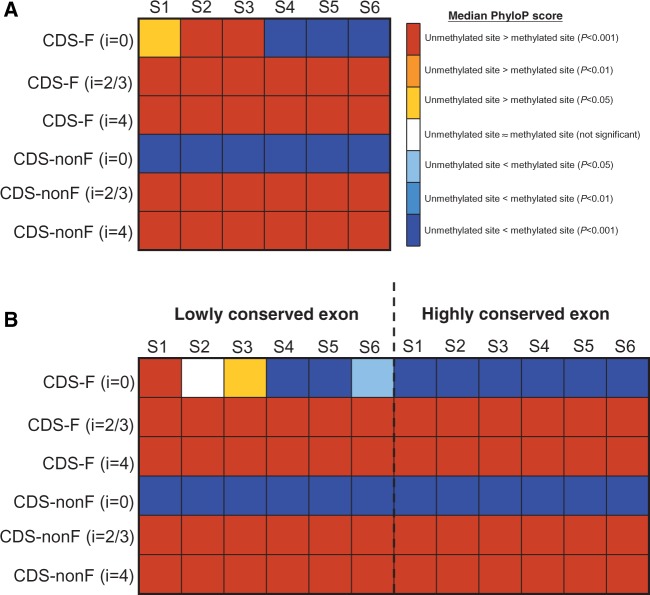
DNA methylation at CpG dinucleotides can significantly increase the rate of cytosine-to-thymine mutations and the level of sequence divergence. Although the correlations between DNA methylation and genomic sequence evolution have been widely studied, an unaddressed yet fundamental question is how DNA methylation is associated with the conservation of individual nucleotides in different sequence contexts. Here, we demonstrate that in mammalian exons, the correlations between DNA methylation and the conservation of individual nucleotides are dependent on the type of exonic sequence (coding or untranslated), the degeneracy of coding nucleotides, background selection pressure, and the relative position (first or nonfirst exon in the transcript) where the nucleotides are located. For untranslated and nonzero-fold degenerate nucleotides, methylated sites are less conserved than unmethylated sites regardless of background selection pressure and the relative position of the exon. For zero-fold degenerate (or nondegenerate) nucleotides, however, the reverse trend is observed in nonfirst coding exons and first coding exons that are under stringent background selection pressure. Furthermore, cytosine-to-thymine mutations at methylated zero-fold degenerate nucleotides are predicted to be more detrimental than those that occur at unmethylated nucleotides. As zero-fold and nonzero-fold degenerate nucleotides are very close to each other, our results suggest that the "functional resolution" of DNA methylation may be finer than previously recognized. In addition, the positive correlation between CpG methylation and the level of conservation at zero-fold degenerate nucleotides implies that CpG methylation may serve as an "indicator" of functional importance of these nucleotides.
Keywords: DNA methylation; degeneracy of nucleotide; genomics; methylation-associated mutation; single-nucleotide evolution.
Figures


Similar articles
-
The evolution of the coding exome of the Arabidopsis species--the influences of DNA methylation, relative exon position, and exon length.BMC Evol Biol. 2014 Jun 25;14:145. doi: 10.1186/1471-2148-14-145. BMC Evol Biol. 2014. PMID: 24965500 Free PMC article.
-
CpG island clusters and pro-epigenetic selection for CpGs in protein-coding exons of HOX and other transcription factors.Proc Natl Acad Sci U S A. 2010 Aug 31;107(35):15485-90. doi: 10.1073/pnas.1010506107. Epub 2010 Aug 17. Proc Natl Acad Sci U S A. 2010. PMID: 20716685 Free PMC article.
-
Position-dependent correlations between DNA methylation and the evolutionary rates of mammalian coding exons.Proc Natl Acad Sci U S A. 2012 Sep 25;109(39):15841-6. doi: 10.1073/pnas.1208214109. Epub 2012 Sep 10. Proc Natl Acad Sci U S A. 2012. PMID: 23019368 Free PMC article.
-
Monitoring methylation changes in cancer.Adv Biochem Eng Biotechnol. 2007;104:1-11. doi: 10.1007/10_024. Adv Biochem Eng Biotechnol. 2007. PMID: 17290816 Review.
-
Study of tissue-specific CpG methylation of DNA in extended genomic loci.Biochemistry (Mosc). 2005 May;70(5):596-603. doi: 10.1007/s10541-005-0153-9. Biochemistry (Mosc). 2005. PMID: 15948713 Review.
Cited by
-
DNA methylation is associated with codon degeneracy in a species of bumblebee.Heredity (Edinb). 2023 Apr;130(4):188-195. doi: 10.1038/s41437-023-00591-z. Epub 2023 Jan 19. Heredity (Edinb). 2023. PMID: 36658299 Free PMC article.
-
The hologenome of Daphnia magna reveals possible DNA methylation and microbiome-mediated evolution of the host genome.Nucleic Acids Res. 2023 Oct 13;51(18):9785-9803. doi: 10.1093/nar/gkad685. Nucleic Acids Res. 2023. PMID: 37638757 Free PMC article.
-
Multi-omics Data Integration for Identifying Osteoporosis Biomarkers and Their Biological Interaction and Causal Mechanisms.iScience. 2020 Feb 21;23(2):100847. doi: 10.1016/j.isci.2020.100847. Epub 2020 Jan 17. iScience. 2020. PMID: 32058959 Free PMC article.
-
Impacts of pretranscriptional DNA methylation, transcriptional transcription factor, and posttranscriptional microRNA regulations on protein evolutionary rate.Genome Biol Evol. 2014 Jun 12;6(6):1530-41. doi: 10.1093/gbe/evu124. Genome Biol Evol. 2014. PMID: 24923326 Free PMC article.
-
Comparative analysis reveals epigenomic evolution related to species traits and genomic imprinting in mammals.Innovation (Camb). 2023 Apr 28;4(3):100434. doi: 10.1016/j.xinn.2023.100434. eCollection 2023 May 15. Innovation (Camb). 2023. PMID: 37215528 Free PMC article.
References
-
- Anastasiadou C, Malousi A, Maglaveras N, Kouidou S. Human epigenome data reveal increased CpG methylation in alternatively spliced sites and putative exonic splicing enhancers. DNA Cell Biol. 2011;30:267–275. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
