

Phylogenetic diversity of the enteric pathogen Salmonella enterica subsp. enterica inferred from genome-wide reference-free SNP characters
- PMID: 24158624
- PMCID: PMC3845640
- DOI: 10.1093/gbe/evt159
Phylogenetic diversity of the enteric pathogen Salmonella enterica subsp. enterica inferred from genome-wide reference-free SNP characters
Abstract
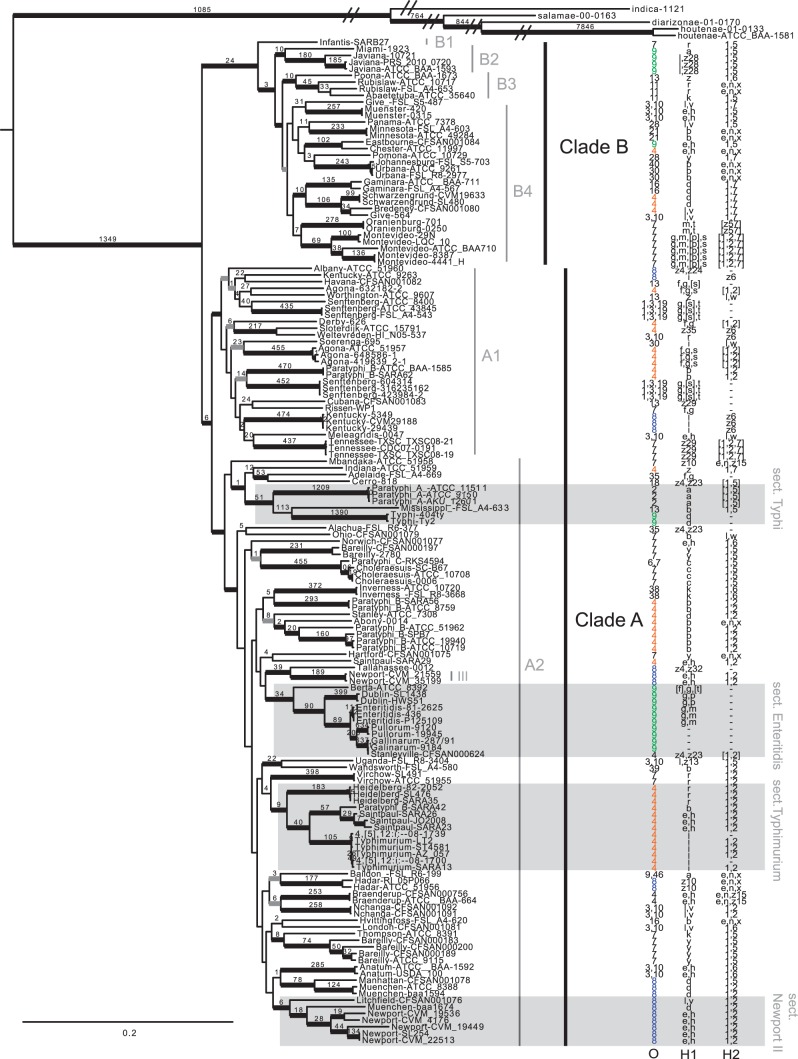
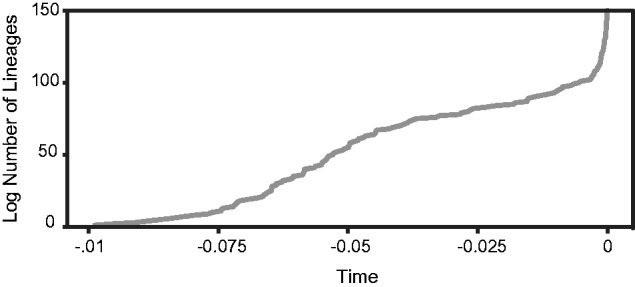
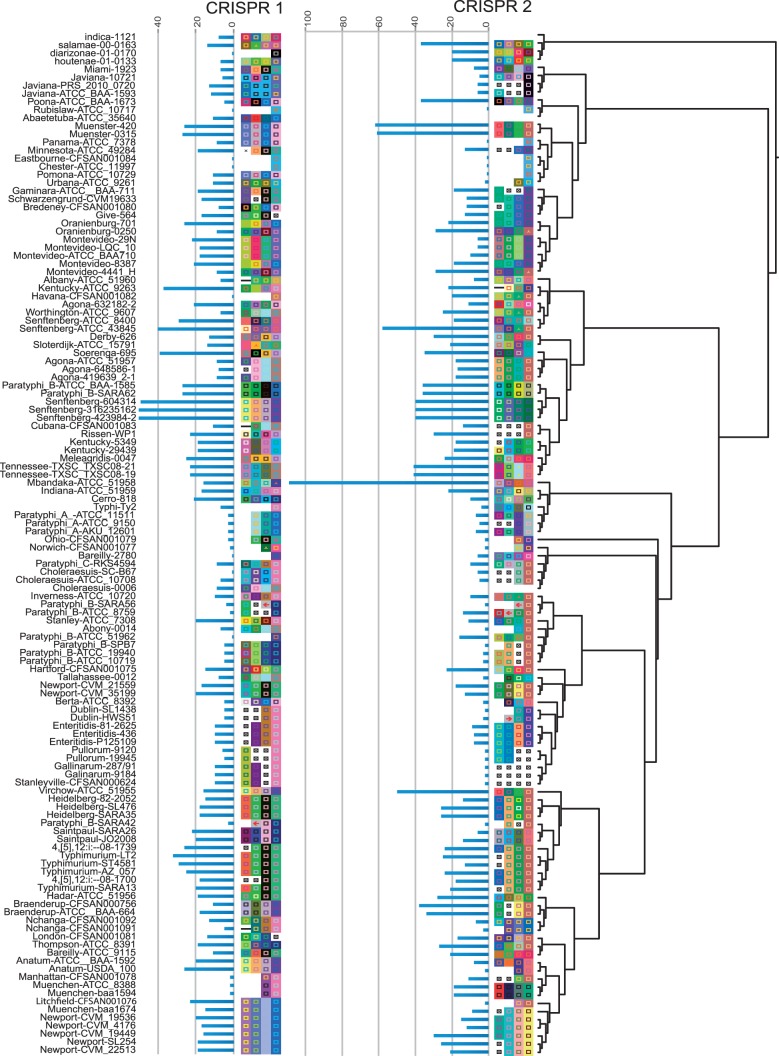
The enteric pathogen Salmonella enterica is one of the leading causes of foodborne illness in the world. The species is extremely diverse, containing more than 2,500 named serovars that are designated for their unique antigen characters and pathogenicity profiles-some are known to be virulent pathogens, while others are not. Questions regarding the evolution of pathogenicity, significance of antigen characters, diversity of clustered regularly interspaced short palindromic repeat (CRISPR) loci, among others, will remain elusive until a strong evolutionary framework is established. We present the first large-scale S. enterica subsp. enterica phylogeny inferred from a new reference-free k-mer approach of gathering single nucleotide polymorphisms (SNPs) from whole genomes. The phylogeny of 156 isolates representing 78 serovars (102 were newly sequenced) reveals two major lineages, each with many strongly supported sublineages. One of these lineages is the S. Typhi group; well nested within the phylogeny. Lineage-through-time analyses suggest there have been two instances of accelerated rates of diversification within the subspecies. We also found that antigen characters and CRISPR loci reveal different evolutionary patterns than that of the phylogeny, suggesting that a horizontal gene transfer or possibly a shared environmental acquisition might have influenced the present character distribution. Our study also shows the ability to extract reference-free SNPs from a large set of genomes and then to use these SNPs for phylogenetic reconstruction. This automated, annotation-free approach is an important step forward for bacterial disease tracking and in efficiently elucidating the evolutionary history of highly clonal organisms.
Keywords: CRISPR; H antigens; O antigens; comparative method; lineage-through-time plot; serovar.
Figures

References
-
- Barker RM, et al. Types of Salmonella Paratyphi B and their phylogenetic significance. J Med Microbiol. 1988;26:285–293. - PubMed
-
- Barrangou R, Horvath P. CRISPR: new horizons in phage resistance and strain identification. Annu Rev Food Sci Technol. 2012;3:143–162. - PubMed
-
- Baum D, Shaw KL. Genealogical perspectives on the species problem. In: Hoch PC, Stephenson AC, editors. Experimental and molecular approaches to plant biosystematics. St. Louis (MO): Missouri Botanical Garden; 1995.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
