

Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects
- PMID: 24158655
- PMCID: PMC3912414
- DOI: 10.1101/gr.158253.113
Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects
Abstract
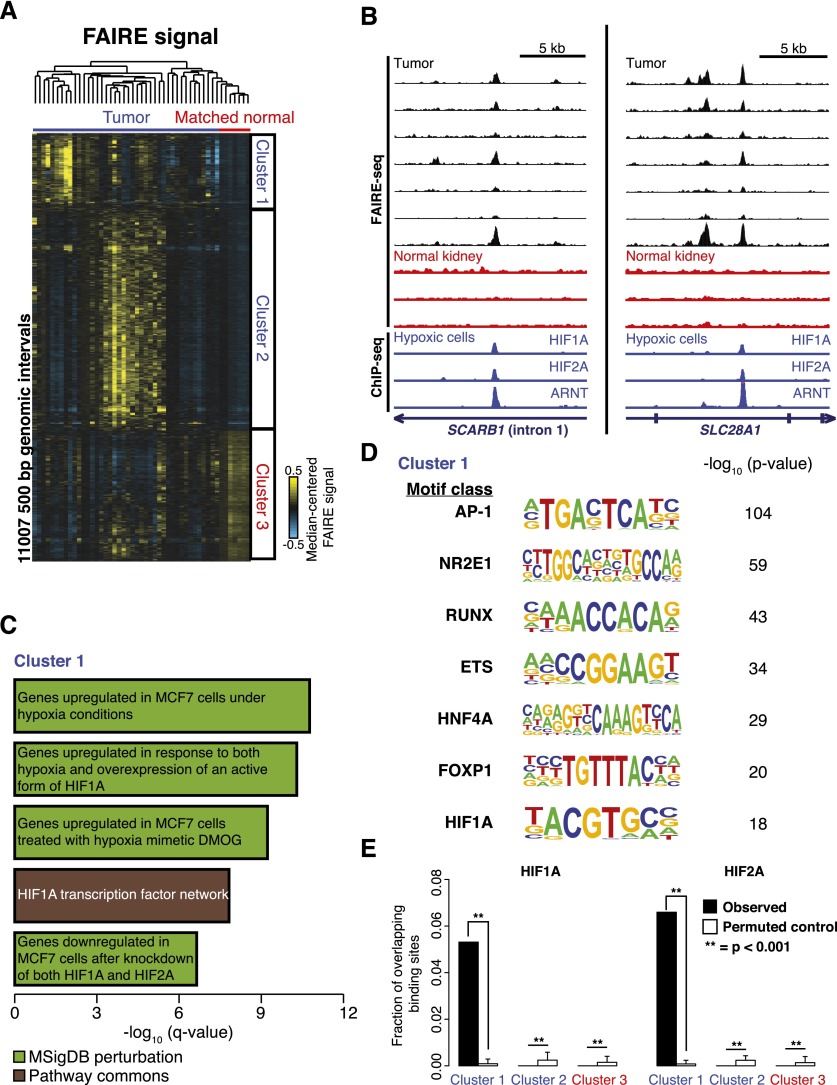
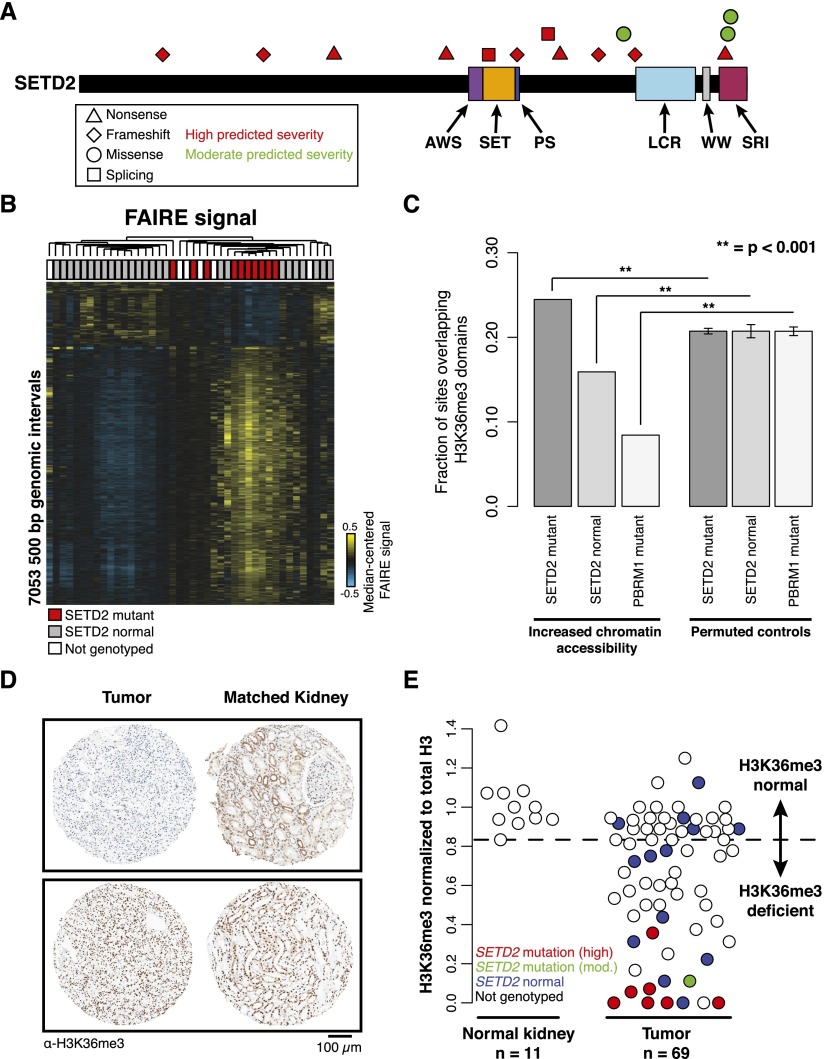
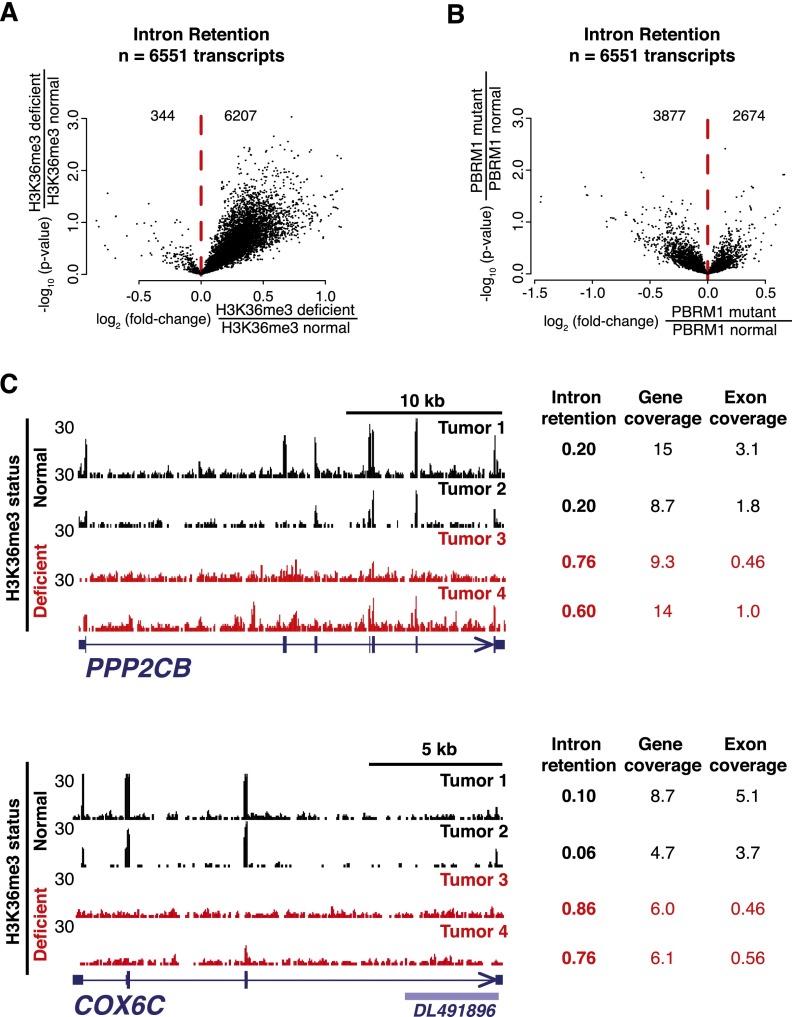
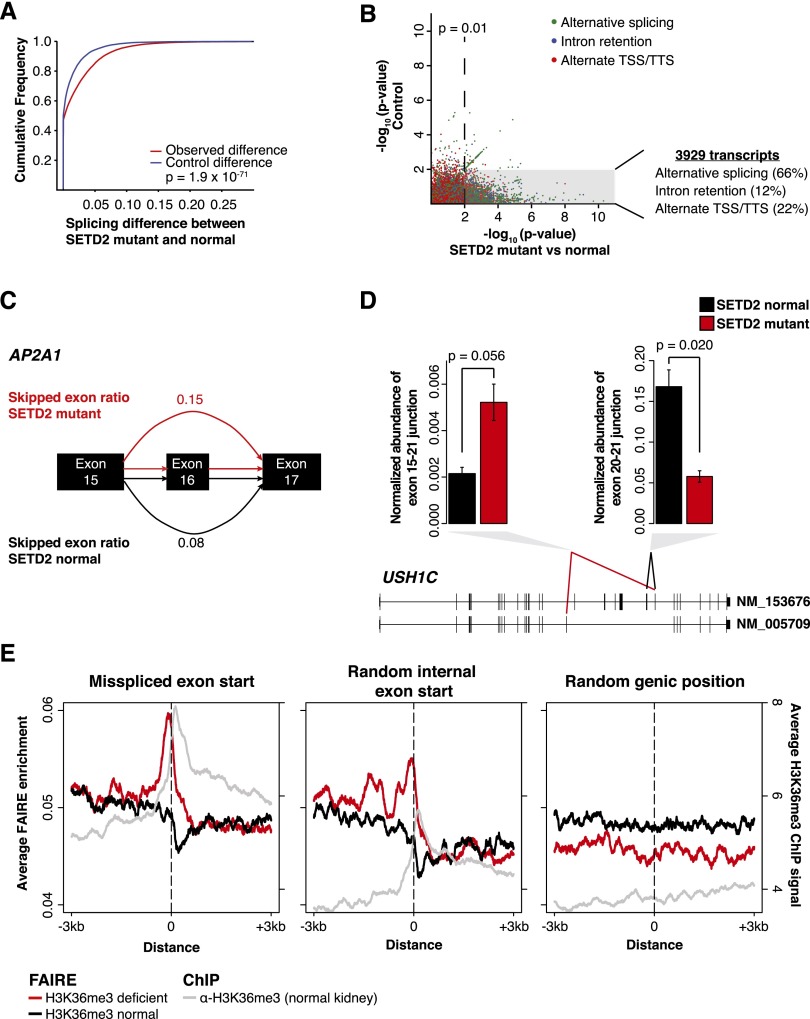
Comprehensive sequencing of human cancers has identified recurrent mutations in genes encoding chromatin regulatory proteins. For clear cell renal cell carcinoma (ccRCC), three of the five commonly mutated genes encode the chromatin regulators PBRM1, SETD2, and BAP1. How these mutations alter the chromatin landscape and transcriptional program in ccRCC or other cancers is not understood. Here, we identified alterations in chromatin organization and transcript profiles associated with mutations in chromatin regulators in a large cohort of primary human kidney tumors. By associating variation in chromatin organization with mutations in SETD2, which encodes the enzyme responsible for H3K36 trimethylation, we found that changes in chromatin accessibility occurred primarily within actively transcribed genes. This increase in chromatin accessibility was linked with widespread alterations in RNA processing, including intron retention and aberrant splicing, affecting ∼25% of all expressed genes. Furthermore, decreased nucleosome occupancy proximal to misspliced exons was observed in tumors lacking H3K36me3. These results directly link mutations in SETD2 to chromatin accessibility changes and RNA processing defects in cancer. Detecting the functional consequences of specific mutations in chromatin regulatory proteins in primary human samples could ultimately inform the therapeutic application of an emerging class of chromatin-targeted compounds.
Figures
References
-
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K 2007. High-resolution profiling of histone methylations in the human genome. Cell 129: 823–837 - PubMed
-
- Batsche E, Yaniv M, Muchardt C 2006. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat Struct Mol Biol 13: 22–29 - PubMed
-
- Bratslavsky G, Sudarshan S, Neckers L, Linehan WM 2007. Pseudohypoxic pathways in renal cell carcinoma. Clin Cancer Res 13: 4667–4671 - PubMed
-
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, et al. 2005. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123: 581–592 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01CA121781/CA/NCI NIH HHS/United States
- T32 GM067553/GM/NIGMS NIH HHS/United States
- P30 CA016086/CA/NCI NIH HHS/United States
- R01CA166447/CA/NCI NIH HHS/United States
- T32 CA071341/CA/NCI NIH HHS/United States
- T32GM08093/GM/NIGMS NIH HHS/United States
- T32 GM007092/GM/NIGMS NIH HHS/United States
- T32 GM008719/GM/NIGMS NIH HHS/United States
- T32GM067553/GM/NIGMS NIH HHS/United States
- R01 GM078994/GM/NIGMS NIH HHS/United States
- 3P30ES010126/ES/NIEHS NIH HHS/United States
- P30 ES010126/ES/NIEHS NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- K12 CA090628/CA/NCI NIH HHS/United States
- R01 CA166447/CA/NCI NIH HHS/United States
- R01 HG006272/HG/NHGRI NIH HHS/United States
- R01 CA121781/CA/NCI NIH HHS/United States
- P30 CA015083/CA/NCI NIH HHS/United States
- T32GM008719/GM/NIGMS NIH HHS/United States
- R01HG006272/HG/NHGRI NIH HHS/United States
- 3P30CA016086/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous