

Atypical hemolytic uremic syndrome
- PMID: 24161037
- PMCID: PMC3863953
- DOI: 10.1016/j.semnephrol.2013.08.003
Atypical hemolytic uremic syndrome
Abstract
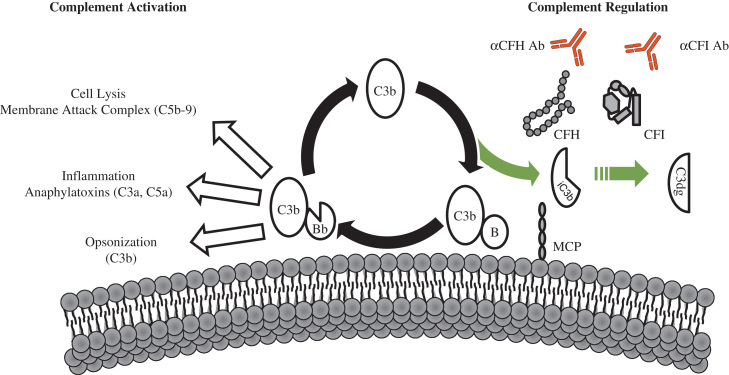
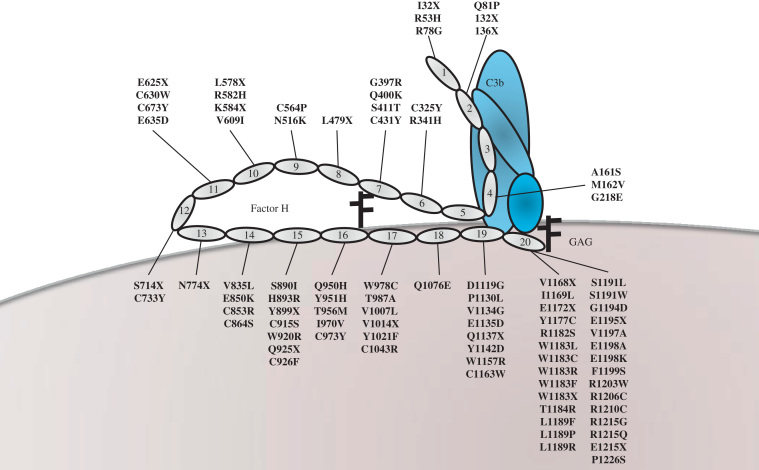
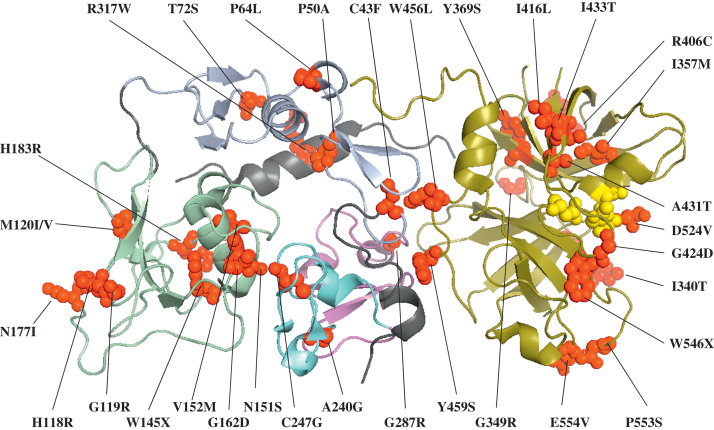
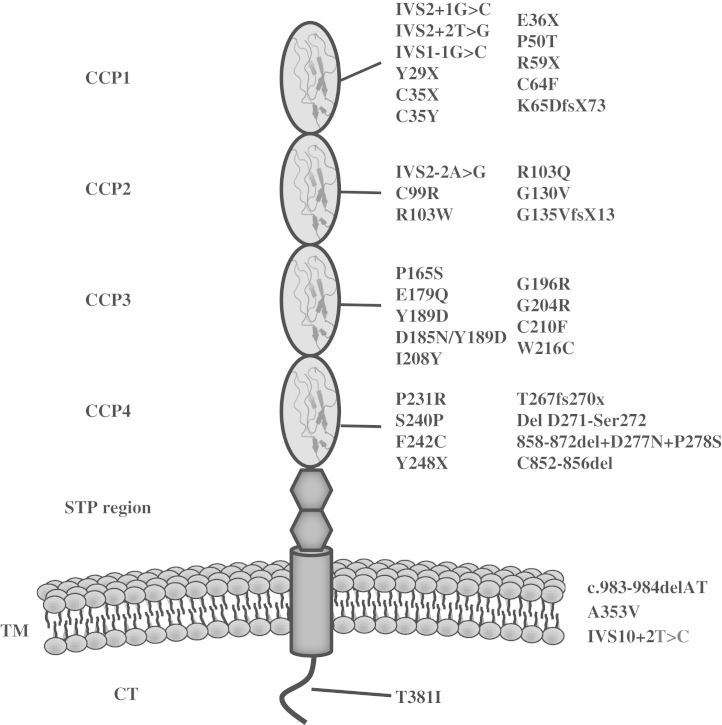
Hemolytic uremic syndrome (HUS) is a triad of microangiopathic hemolytic anemia, thrombocytopenia, and acute renal failure. The atypical form of HUS is a disease characterized by complement overactivation. Inherited defects in complement genes and acquired autoantibodies against complement regulatory proteins have been described. Incomplete penetrance of mutations in all predisposing genes is reported, suggesting that a precipitating event or trigger is required to unmask the complement regulatory deficiency. The underlying genetic defect predicts the prognosis both in native kidneys and after renal transplantation. The successful trials of the complement inhibitor eculizumab in the treatment of atypical HUS will revolutionize disease management.
Keywords: Complement; eculizumab; factor H; factor I; hemolytic uremic syndrome; membrane cofactor protein; thrombomodulin; transplantation.
© 2013 Elsevier Inc. All rights reserved.
Figures
References
-
- Kavanagh D., Richards A., Atkinson J. Complement regulatory genes and hemolytic uremic syndromes. Annu Rev Med. 2008;59:293–309. - PubMed
-
- Besbas N., Karpman D., Landau D., Loirat C., Proesmans W., Remuzzi G. A classification of hemolytic uremic syndrome and thrombotic thrombocytopenic purpura and related disorders. Kidney Int. 2006;70:423–431. - PubMed
-
- Constantinescu A.R., Bitzan M., Weiss L.S., Christen E., Kaplan B.S., Cnaan A. Non-enteropathic hemolytic uremic syndrome: causes and short-term course. Am J Kidney Dis. 2004;43:976–982. - PubMed
-
- Fogo A., Kashgarian M. Elsevier Science; Amsterdam: 2005. Diagnostic atlas of renal pathology.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
