

Mitochondrial oxidative stress is the Achille's heel of melanoma cells resistant to Braf-mutant inhibitor
- PMID: 24161908
- PMCID: PMC3875764
- DOI: 10.18632/oncotarget.1420
Mitochondrial oxidative stress is the Achille's heel of melanoma cells resistant to Braf-mutant inhibitor
Abstract
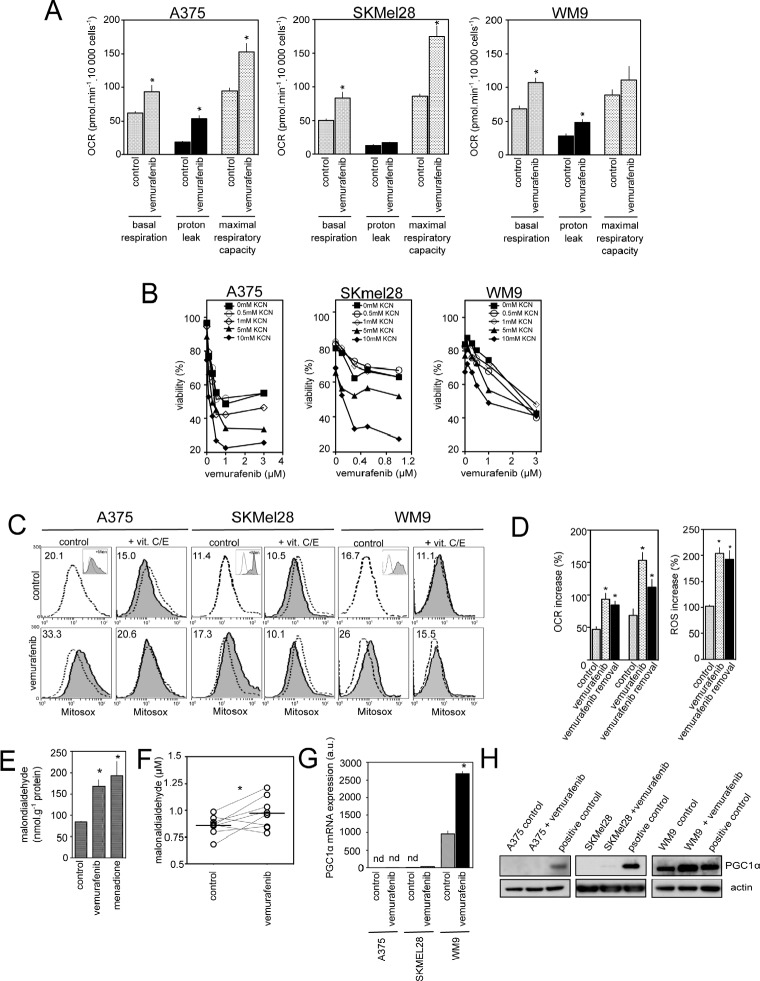
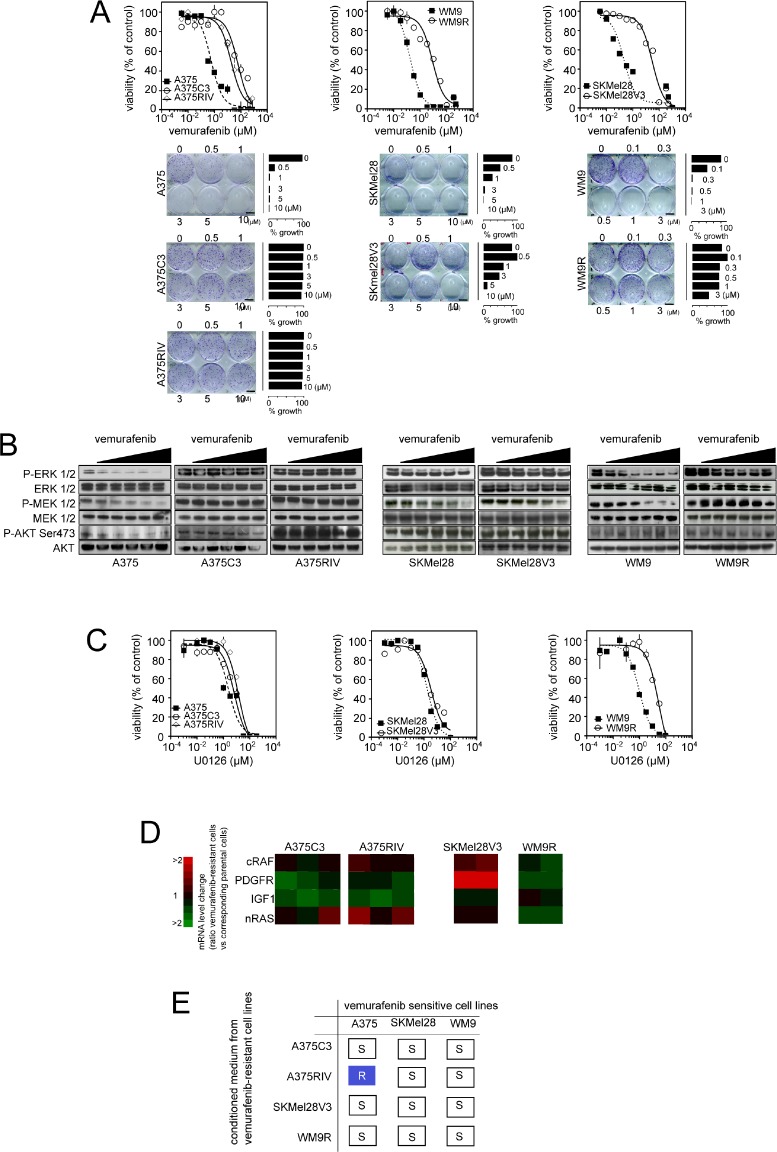
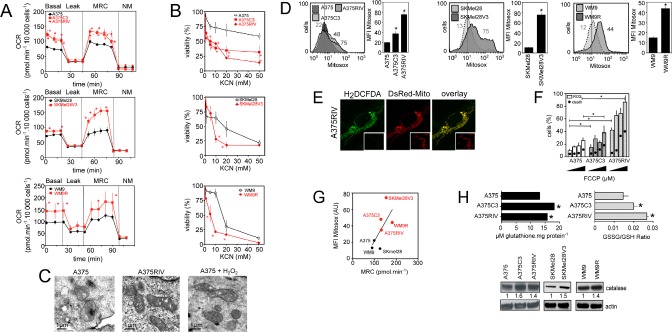
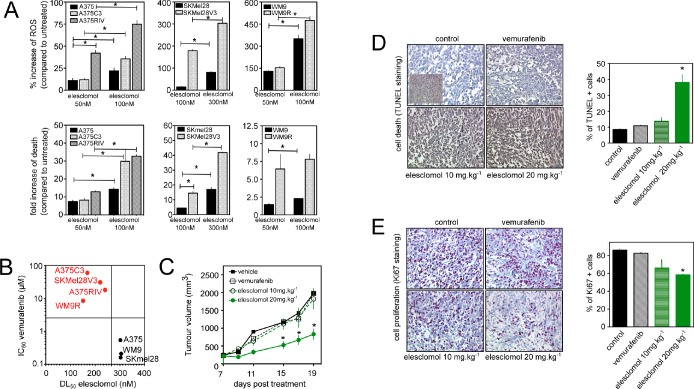
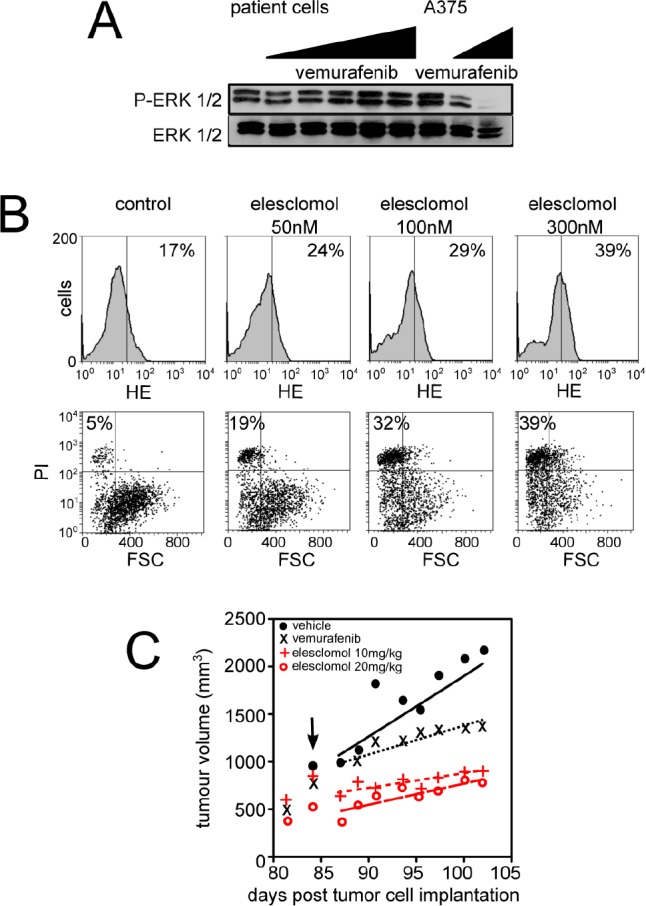
Vemurafenib/PLX4032, a selective inhibitor of mutant BRAFV600E, constitutes a paradigm shift in melanoma therapy. Unfortunately, acquired resistance, which unavoidably occurs, represents one major limitation to clinical responses. Recent studies have highlighted that vemurafenib activated oxidative metabolism in BRAFV600E melanomas expressing PGC1α. However, the oxidative state of melanoma resistant to BRAF inhibitors is unknown. We established representative in vitro and in vivo models of human melanoma resistant to vemurafenib including primary specimens derived from melanoma patients. Firstly, our study reveals that vemurafenib increased mitochondrial respiration and ROS production in BRAFV600E melanoma cell lines regardless the expression of PGC1α. Secondly, melanoma cells that have acquired resistance to vemurafenib displayed intrinsically high rates of mitochondrial respiration associated with elevated mitochondrial oxidative stress irrespective of the presence of vemurafenib. Thirdly, the elevated ROS level rendered vemurafenib-resistant melanoma cells prone to cell death induced by pro-oxidants including the clinical trial drug, elesclomol. Based on these observations, we propose that the mitochondrial oxidative signature of resistant melanoma constitutes a novel opportunity to overcome resistance to BRAF inhibition.
Conflict of interest statement
Dr. Ariel Savina is employed by Roche SAS, which provided a support to this work. Vemurafenib was synthesized by Roche SAS.
Figures
References
-
- Dong J, Phelps RG, Qiao R, Yao S, Benard O, Ronai Z, Aaronson SA. BRAF oncogenic mutations correlate with progression rather than initiation of human melanoma. Cancer Res. 2003;63:3883–5. - PubMed
-
- Falchook GS, Trent JC, Heinrich MC, Beadling C, Patterson J, Bastida CC, Blackman SC, Kurzrock R. BRAF mutant gastrointestinal stromal tumor: first report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget. 2013;4:310–5. - PMC - PubMed
-
- Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, Hersey P, Kefford R, Lawrence D, Puzanov I, Lewis KD, Amaravadi RK, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012;366:707–14. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
