

Improved exome prioritization of disease genes through cross-species phenotype comparison
- PMID: 24162188
- PMCID: PMC3912424
- DOI: 10.1101/gr.160325.113
Improved exome prioritization of disease genes through cross-species phenotype comparison
Abstract
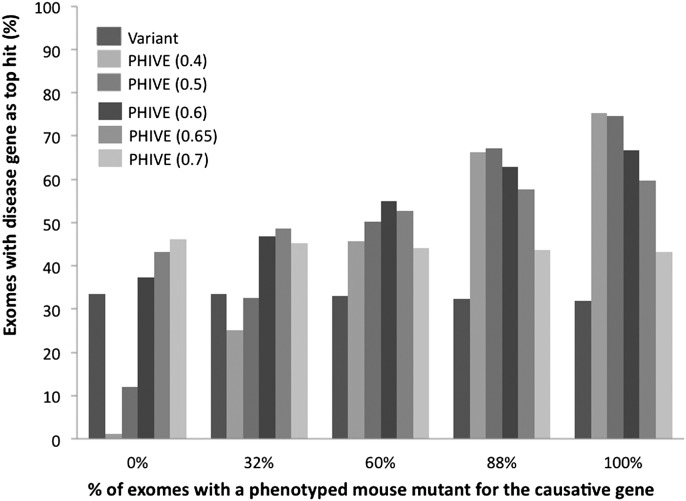
Numerous new disease-gene associations have been identified by whole-exome sequencing studies in the last few years. However, many cases remain unsolved due to the sheer number of candidate variants remaining after common filtering strategies such as removing low quality and common variants and those deemed unlikely to be pathogenic. The observation that each of our genomes contains about 100 genuine loss-of-function variants makes identification of the causative mutation problematic when using these strategies alone. We propose using the wealth of genotype to phenotype data that already exists from model organism studies to assess the potential impact of these exome variants. Here, we introduce PHenotypic Interpretation of Variants in Exomes (PHIVE), an algorithm that integrates the calculation of phenotype similarity between human diseases and genetically modified mouse models with evaluation of the variants according to allele frequency, pathogenicity, and mode of inheritance approaches in our Exomiser tool. Large-scale validation of PHIVE analysis using 100,000 exomes containing known mutations demonstrated a substantial improvement (up to 54.1-fold) over purely variant-based (frequency and pathogenicity) methods with the correct gene recalled as the top hit in up to 83% of samples, corresponding to an area under the ROC curve of >95%. We conclude that incorporation of phenotype data can play a vital role in translational bioinformatics and propose that exome sequencing projects should systematically capture clinical phenotypes to take advantage of the strategy presented here.
Figures




References
-
- Aerts S, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, Tranchevent LC, De Moor B, Marynen P, Hassan B, et al. 2006. Gene prioritization through genomic data fusion. Nat Biotechnol 24: 537–544 - PubMed
-
- Amberger J, Bocchini C, Hamosh A 2011. A new face and new challenges for Online Mendelian Inheritance in Man (OMIM®). Hum Mutat 32: 564–567 - PubMed
-
- Ayadi A, Birling MC, Bottomley J, Bussell J, Fuchs H, Fray M, Gailus-Durner V, Greenaway S, Houghton R, Karp N, et al. 2012. Mouse large-scale phenotyping initiatives: Overview of the European Mouse Disease Clinic (EUMODIC) and of the Wellcome Trust Sanger Institute Mouse Genetics Project. Mamm Genome 23: 600–610 - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources