

Split-hand/foot malformation - molecular cause and implications in genetic counseling
- PMID: 24163146
- PMCID: PMC3909621
- DOI: 10.1007/s13353-013-0178-5
Split-hand/foot malformation - molecular cause and implications in genetic counseling
Abstract
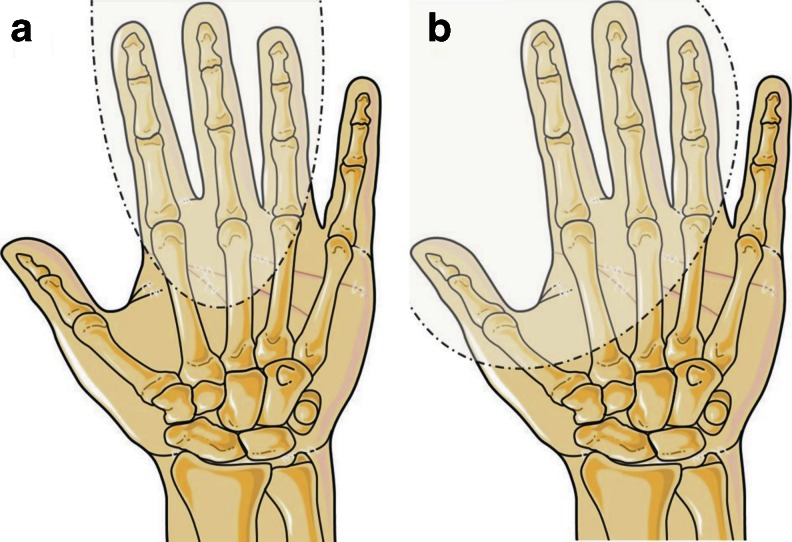
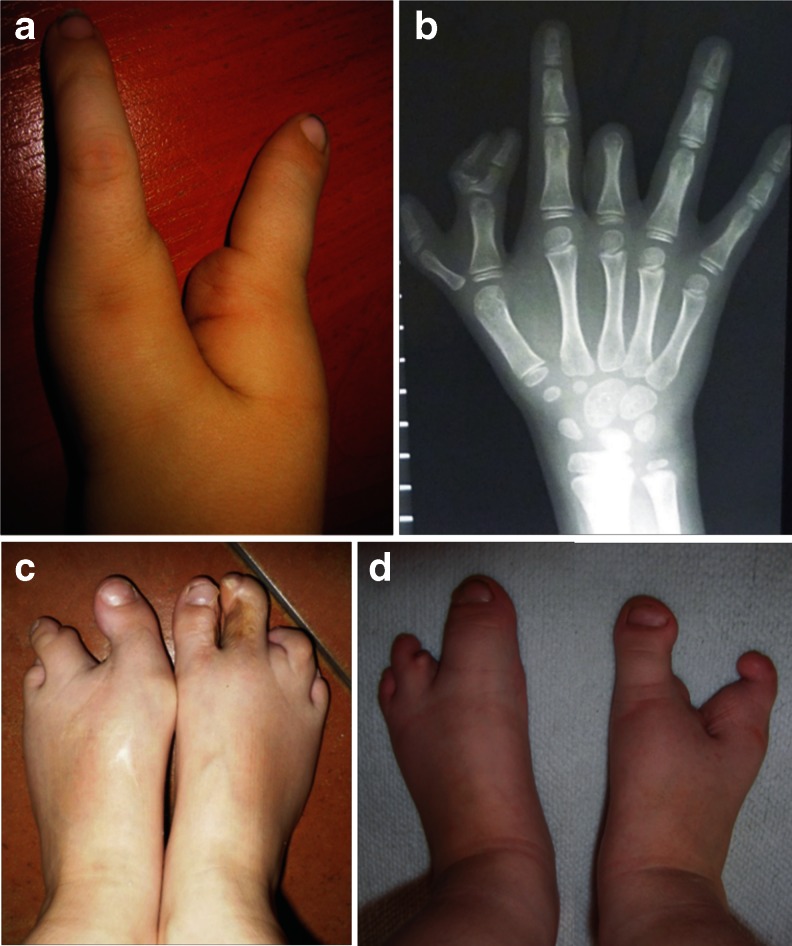
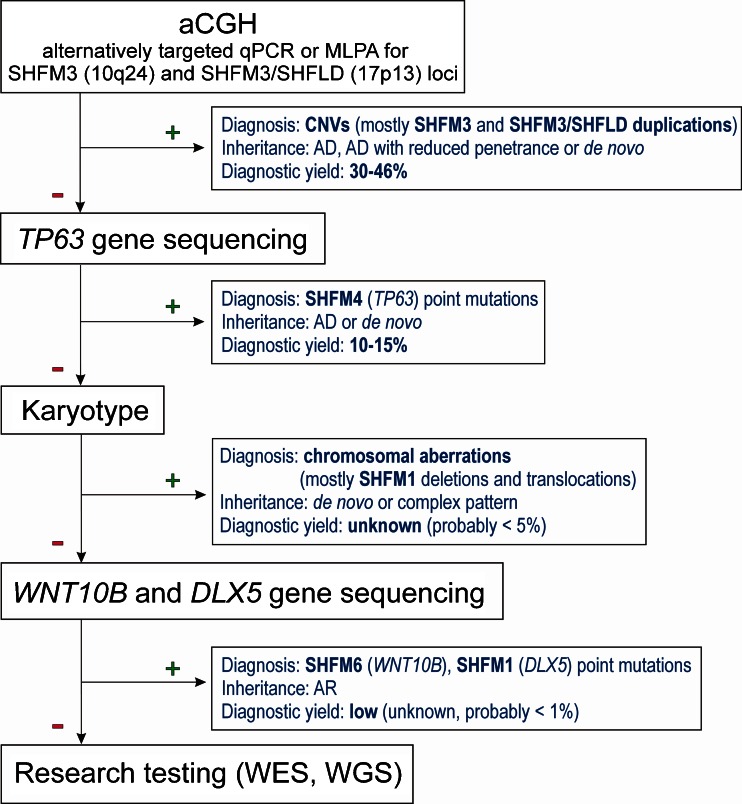
Split-hand/foot malformation (SHFM) is a congenital limb defect affecting predominantly the central rays of the autopod and occurs either as an isolated trait or part of a multiple congenital anomaly syndrome. SHFM is usually sporadic, familial forms are uncommon. The condition is clinically and genetically heterogeneous and shows mostly autosomal dominant inheritance with variable expressivity and reduced penetrance. To date, seven chromosomal loci associated with isolated SHFM have been described, i.e., SHFM1 to 6 and SHFM/SHFLD. The autosomal dominant mode of inheritance is typical for SHFM1, SHFM3, SHFM4, SHFM5. Autosomal recessive and X-linked inheritance is very uncommon and have been noted only in a few families. Most of the known SHFM loci are associated with chromosomal rearrangements that involve small deletions or duplications of the human genome. In addition, three genes, i.e., TP63, WNT10B, and DLX5 are known to carry point mutations in patients affected by SHFM. In this review, we focus on the known molecular basis of isolated SHFM. We provide clinical and molecular information about each type of abnormality as well as discuss the underlying pathways and mechanism that contribute to their development. Recent progress in the understanding of SHFM pathogenesis currently allows for the identification of causative genetic changes in about 50 % of the patients affected by this condition. Therefore, we propose a diagnostic flow-chart helpful in the planning of molecular genetic tests aimed at identifying disease causing mutation. Finally, we address the issue of genetic counseling, which can be extremely difficult and challenging especially in sporadic SHFM cases.
Figures
References
-
- Acampora D, Merlo GR, Paleari L, Zerega B, Postiglione MP, Mantero S, Bober E, Barbieri O, Simeone A, Levi G. Craniofacial, vestibular and bone defects in mice lacking the Distal-less-related gene Dlx5. Development. 1999;126(17):3795–3809. - PubMed
-
- Aten E, den Hollander N, Ruivenkamp C, Knijnenburg J, van Bokhoven H, den Dunnen J. Split hand-foot malformation, tetralogy of Fallot, mental retardation and a 1 Mb 19p deletion – evidence for further heterogeneity? Am J Med Genet A. 2008;152A(8):2053–2056. - PubMed
-
- Bens S, Haake A, Tönnies H, Vater I, Stephani U, Holterhus PM, Siebert R, Caliebe A. A de novo 1.1Mb microdeletion of chromosome 19p13.11 provides indirect evidence for EPS15L1 to be a strong candidate for split hand split foot malformation. Eur J Med Genet. 2011;54(5):e501–e504. doi: 10.1016/j.ejmg.2011.05.004. - DOI - PubMed
-
- Birnbaum RY, Clowney EJ, Agamy O, Kim MJ, Zhao J, Yamanaka T, Pappalardo Z, Clarke SL, Wenger AM, Nguyen L, Gurrieri F, Everman DB, Schwartz CE, Birk OS, Bejerano G, Lomvardas S, Ahituv N. Coding exons function as tissue-specific enhancers of nearby genes. Genome Res. 2012;22:1059–1068. doi: 10.1101/gr.133546.111. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
