

Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites
- PMID: 24163245
- PMCID: PMC3919014
- DOI: 10.1093/hmg/ddt531
Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites
Abstract
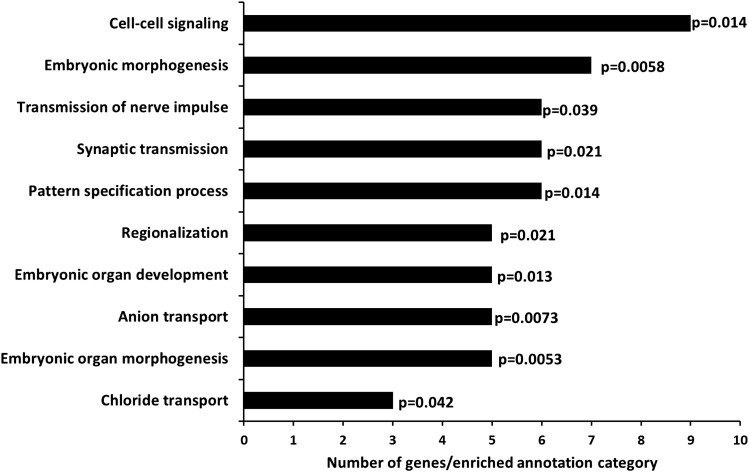
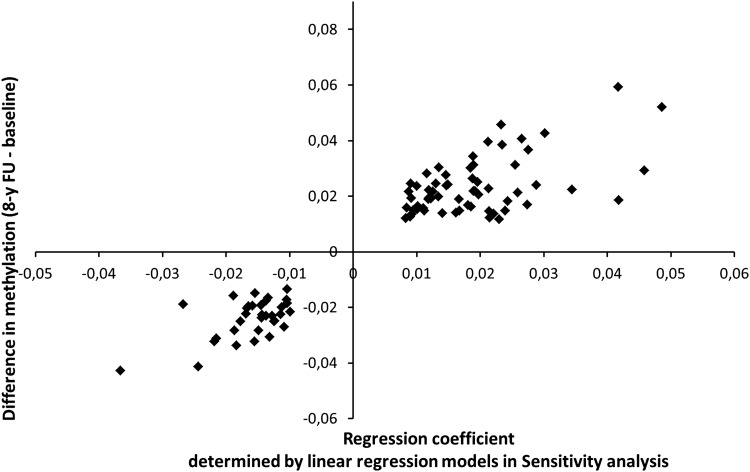
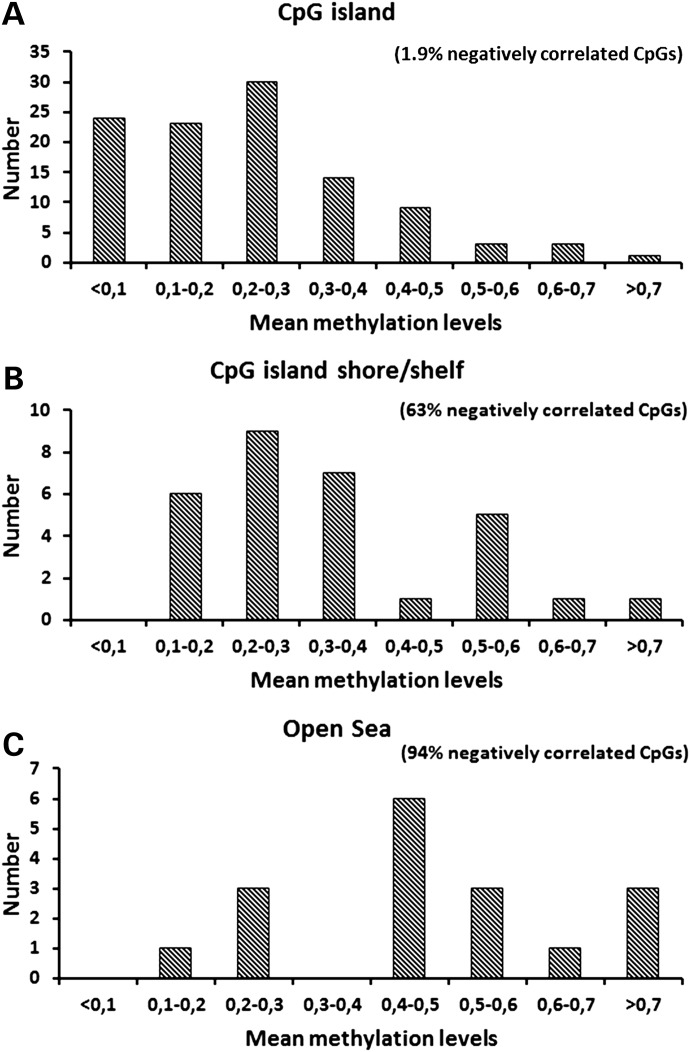
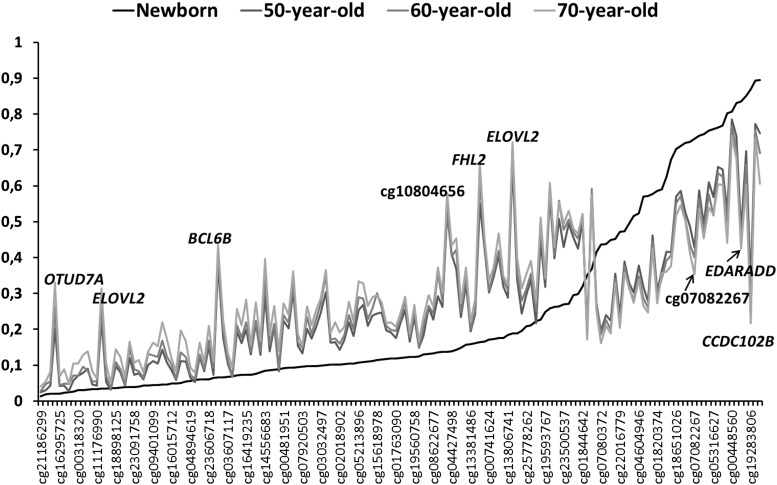
Understanding the role of epigenetic modifications, e.g. DNA methylation, in the process of aging requires the characterization of methylation patterns in large cohorts. We analysed >480 000 CpG sites using Infinium HumanMethylation450 BeadChip (Illumina) in whole blood DNA of 965 participants of a population-based cohort study aged between 50 and 75 years. In an exploratory analysis in 400 individuals, 200 CpG sites with the highest Spearman correlation coefficients for the association between methylation and age were identified. Of these 200 CpGs, 162 were significantly associated with age, which was verified in an independent cohort of 498 individuals using mixed linear regression models adjusted for gender, smoking behaviour, age-related diseases and random batch effect and corrected for multiple testing by Bonferroni. In another independent cohort of 67 individuals without history of major age-related diseases and with a follow-up of 8 years, we observed a gain in methylation at 96% (52%, significant) of the positively age-associated CpGs and a loss at all (89%, significant) of the negatively age-associated CpGs in each individual while getting 8 years older. A regression model for age prediction based on 17 CpGs as predicting variables explained 71% of the variance in age with an average accuracy of 2.6 years. In comparison with cord blood samples obtained from the Ulm Birth Cohort Study, we observed a more than 2-fold change in mean methylation levels from birth to older age at 86 CpGs. We were able to identify 65 novel CpG sites with significant association of methylation with age.
Figures


References
-
- Talens R.P., Jukema J.W., Trompet S., Kremer D., Westendorp R.G., Lumey L.H., Sattar N., Putter H., Slagboom P.E., Heijmans B.T. Hypermethylation at loci sensitive to the prenatal environment is associated with increased incidence of myocardial infarction. Int. J. Epidemiol. 2012;41:106–115. - PMC - PubMed
-
- Langevin S.M., Koestler D.C., Christensen B.C., Butler R.A., Wiencke J.K., Nelson H.H., Houseman E.A., Marsit C.J., Kelsey K.T. Peripheral blood DNA methylation profiles are indicative of head and neck squamous cell carcinoma: an epigenome-wide association study. Epigenetics. 2012;7:291–299. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
