IMG 4 version of the integrated microbial genomes comparative analysis system
- PMID: 24165883
- PMCID: PMC3965111
- DOI: 10.1093/nar/gkt963
IMG 4 version of the integrated microbial genomes comparative analysis system
Abstract
The Integrated Microbial Genomes (IMG) data warehouse integrates genomes from all three domains of life, as well as plasmids, viruses and genome fragments. IMG provides tools for analyzing and reviewing the structural and functional annotations of genomes in a comparative context. IMG's data content and analytical capabilities have increased continuously since its first version released in 2005. Since the last report published in the 2012 NAR Database Issue, IMG's annotation and data integration pipelines have evolved while new tools have been added for recording and analyzing single cell genomes, RNA Seq and biosynthetic cluster data. Different IMG datamarts provide support for the analysis of publicly available genomes (IMG/W: http://img.jgi.doe.gov/w), expert review of genome annotations (IMG/ER: http://img.jgi.doe.gov/er) and teaching and training in the area of microbial genome analysis (IMG/EDU: http://img.jgi.doe.gov/edu).
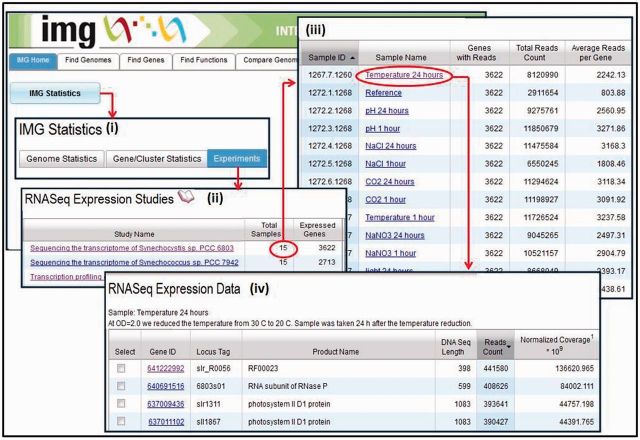
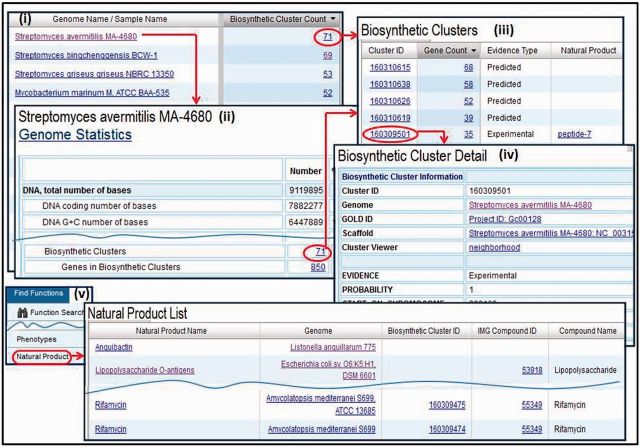
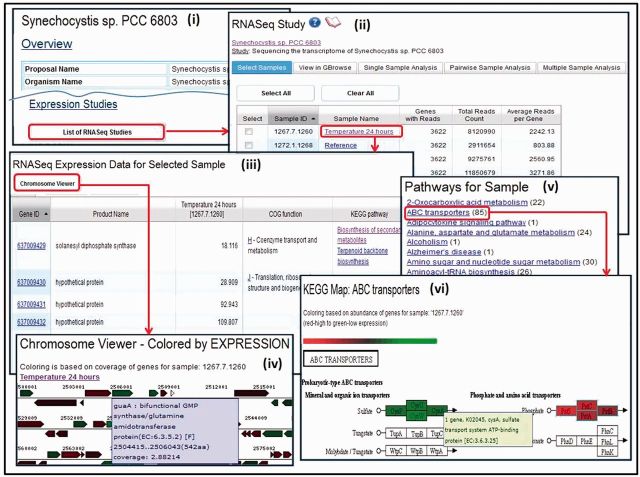
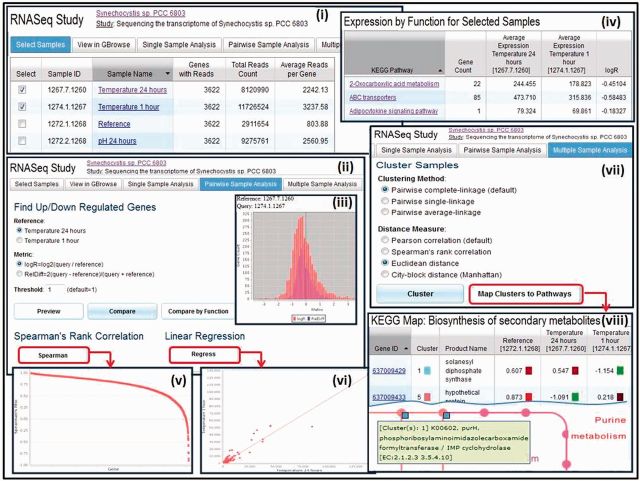
Figures