

Multiple Aspects of Gene Dysregulation in Huntington's Disease
- PMID: 24167500
- PMCID: PMC3806340
- DOI: 10.3389/fneur.2013.00127
Multiple Aspects of Gene Dysregulation in Huntington's Disease
Abstract
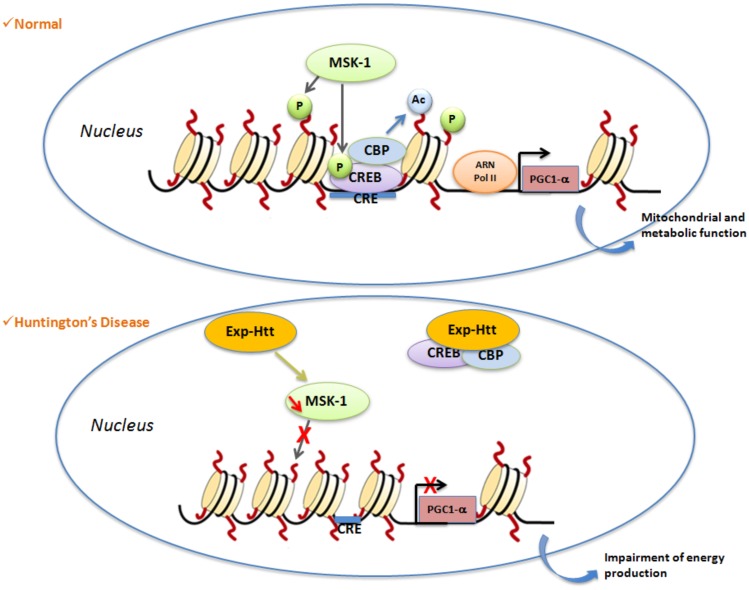
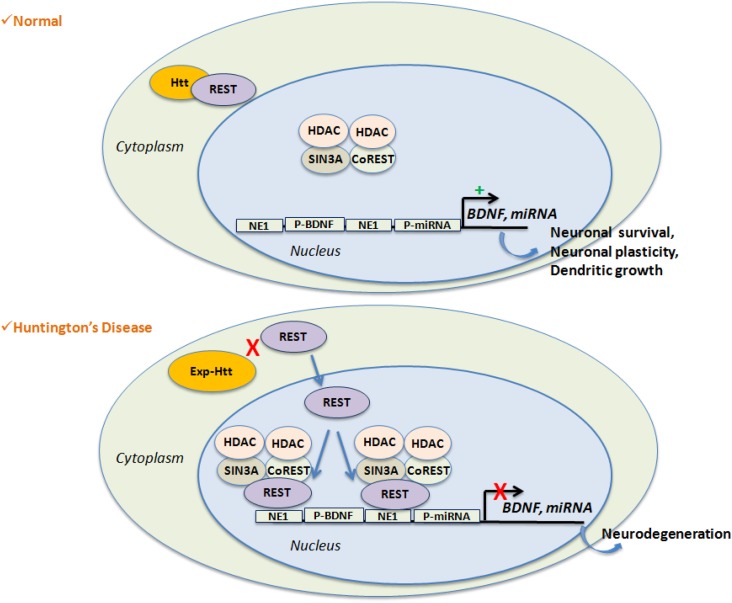
Huntington's Disease (HD) is a genetic neurodegenerative disease caused by a CAG expansion in the gene encoding Huntingtin (Htt). It is characterized by chorea, cognitive, and psychiatric disorders. The most affected brain region is the striatum, and the clinical symptoms are directly correlated to the rate of striatal degeneration. The wild-type Htt is a ubiquitous protein and its deletion is lethal. Mutated (expanded) Htt produces excitotoxicity, mitochondrial dysfunctions, axonal transport deficit, altered proteasome activity, and gene dysregulation. Transcriptional dysregulation occurs at early neuropathological stages in HD patients. Multiple genes are dysregulated, with overlaps of altered transcripts between mouse models of HD and patient brains. Nuclear localization of Exp-Htt interferes with transcription factors, co-activators, and proteins of the transcriptional machinery. Another key mechanism described so far, is an alteration of cytoplasmic retention of the transcriptional repressor REST, which is normally associated with wild-type Htt. As such, Exp-Htt causes alteration of transcription of multiple genes involved in neuronal survival, plasticity, signaling, and mitochondrial biogenesis and respiration. Besides these transcriptional dysregulations, Exp-Htt affects the chromatin structure through altered post-translational modifications (PTM) of histones and methylation of DNA. Multiple alterations of histone PTM are described, including acetylation, methylation, ubiquitylation, polyamination, and phosphorylation. Exp-Htt also affects the expression and regulation of non-coding microRNAs (miRNAs). First multiple neural miRNAs are controlled by REST, and dysregulated in HD, with concomitant de-repression of downstream mRNA targets. Second, Exp-Htt protein or RNA may also play a major role in the processing of miRNAs and hence pathogenesis. These pleiotropic effects of Exp-Htt on gene expression may represent seminal deleterious effects in the pathogenesis of HD.
Keywords: REST; chromatin remodeling; epigenetics; histone modifications; miRNAs; transcription.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials