

EBV finds a polycomb-mediated, epigenetic solution to the problem of oncogenic stress responses triggered by infection
- PMID: 24167519
- PMCID: PMC3807040
- DOI: 10.3389/fgene.2013.00212
EBV finds a polycomb-mediated, epigenetic solution to the problem of oncogenic stress responses triggered by infection
Abstract
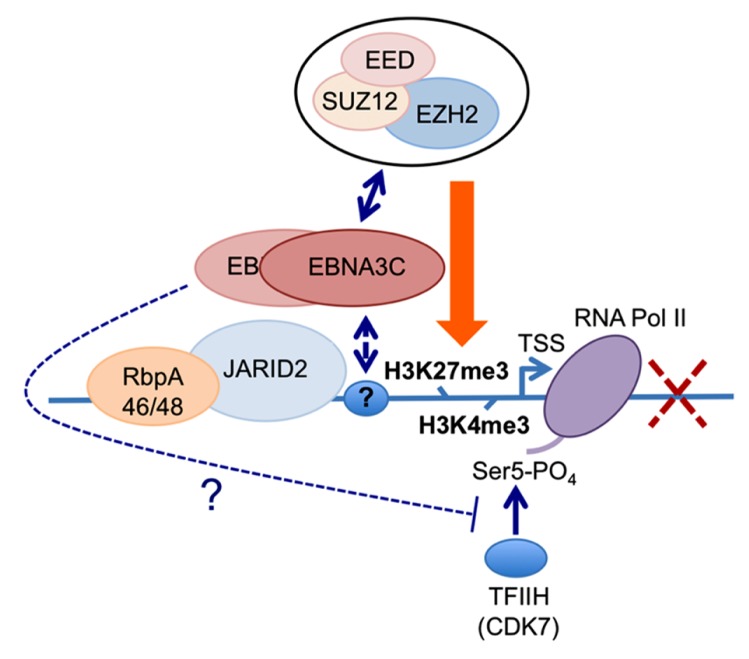
Viruses that establish a persistent infection, involving intracellular latency, commonly stimulate cellular DNA synthesis and sometimes cell division early after infection. However, most cells of metazoans have evolved "fail-safe" responses that normally monitor unscheduled DNA synthesis and prevent cell proliferation when, for instance, cell proto-oncogenes are "activated" by mutation, amplification, or chromosomal rearrangements. These cell intrinsic defense mechanisms that reduce the risk of neoplasia and cancer are collectively called oncogenic stress responses (OSRs). Mechanisms include the activation of tumor suppressor genes and the so-called DNA damage response that together trigger pathways leading to cell cycle arrest (e.g., cell senescence) or complete elimination of cells (e.g., apoptosis). It is not surprising that viruses that can induce cellular DNA synthesis and cell division have the capacity to trigger OSR, nor is it surprising that these viruses have evolved countermeasures for inactivating or bypassing OSR. The main focus of this review is how the human tumor-associated Epstein-Barr virus manipulates the host polycomb group protein system to control - by epigenetic repression of transcription - key components of the OSR during the transformation of normal human B cells into permanent cell lines.
Keywords: B cell transformation; BIM; Epstein–Barr virus; PcG; epigenetic; oncogene-induced senescence; oncogenic stress response; p16INK4a.
Figures



References
-
- Askew D. S., Ashmun R. A., Simmons B. C., Cleveland J. L. (1991). Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene 6 1915–1922 - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources