

Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability to ischemic cell death
- PMID: 24174660
- PMCID: PMC6618359
- DOI: 10.1523/JNEUROSCI.1729-13.2013
Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability to ischemic cell death
Abstract
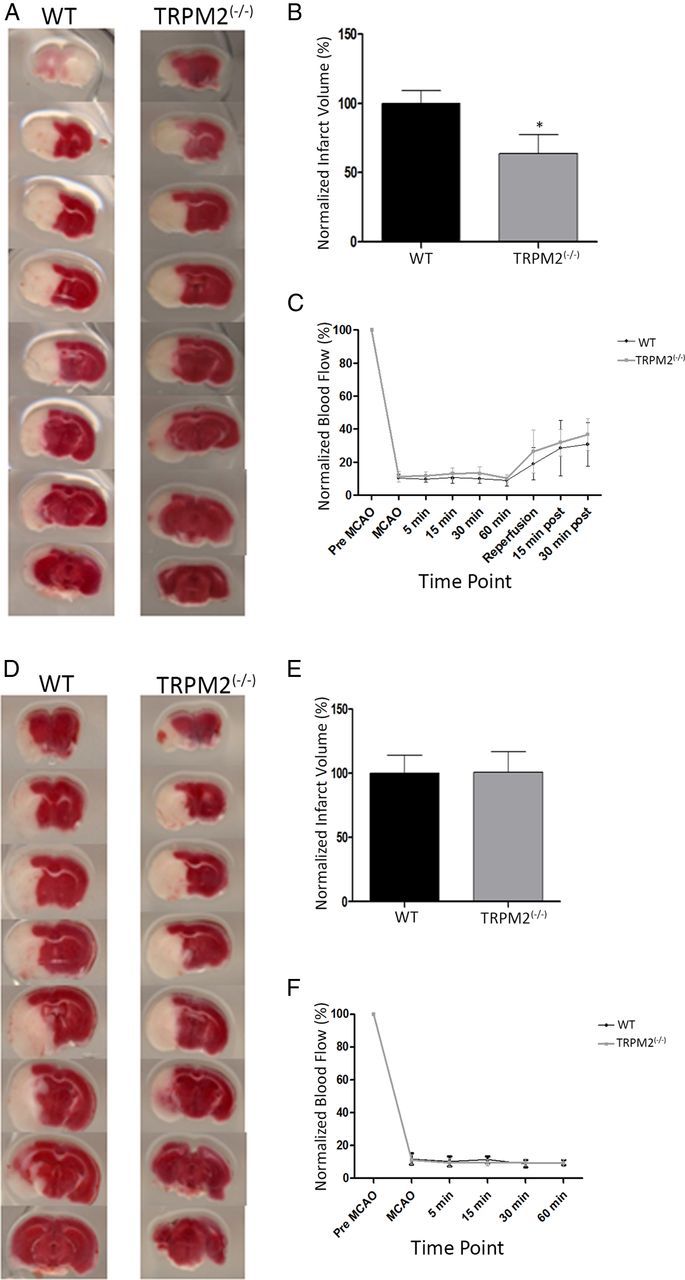
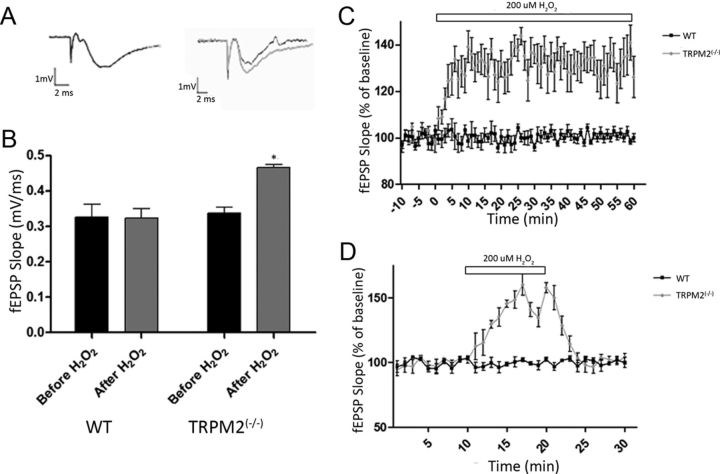
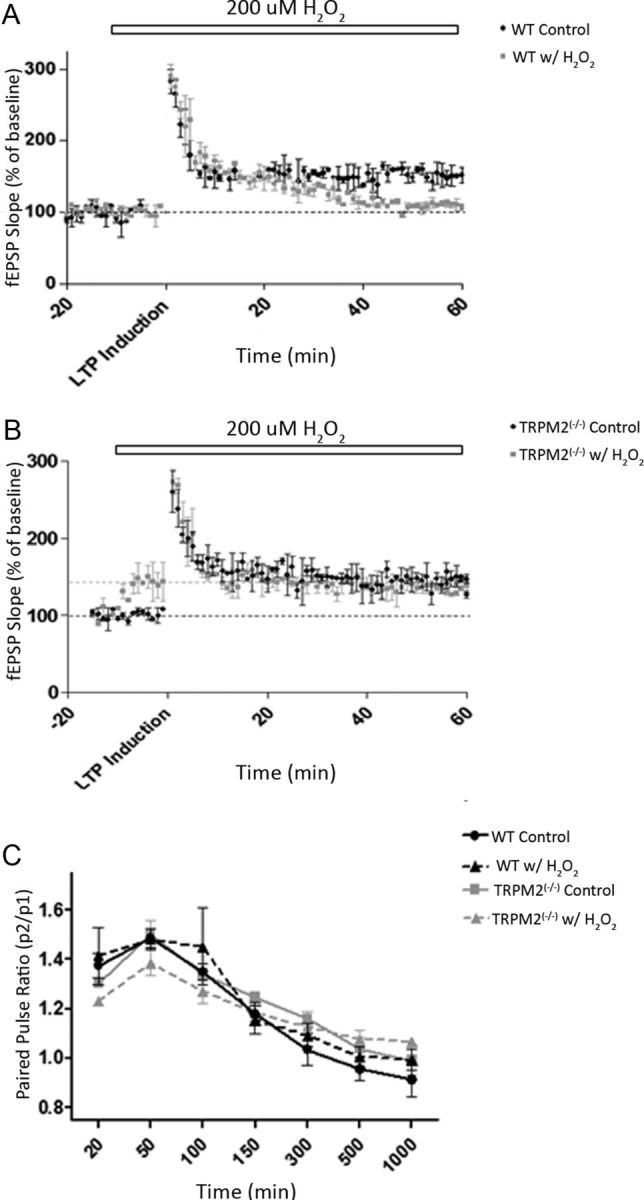
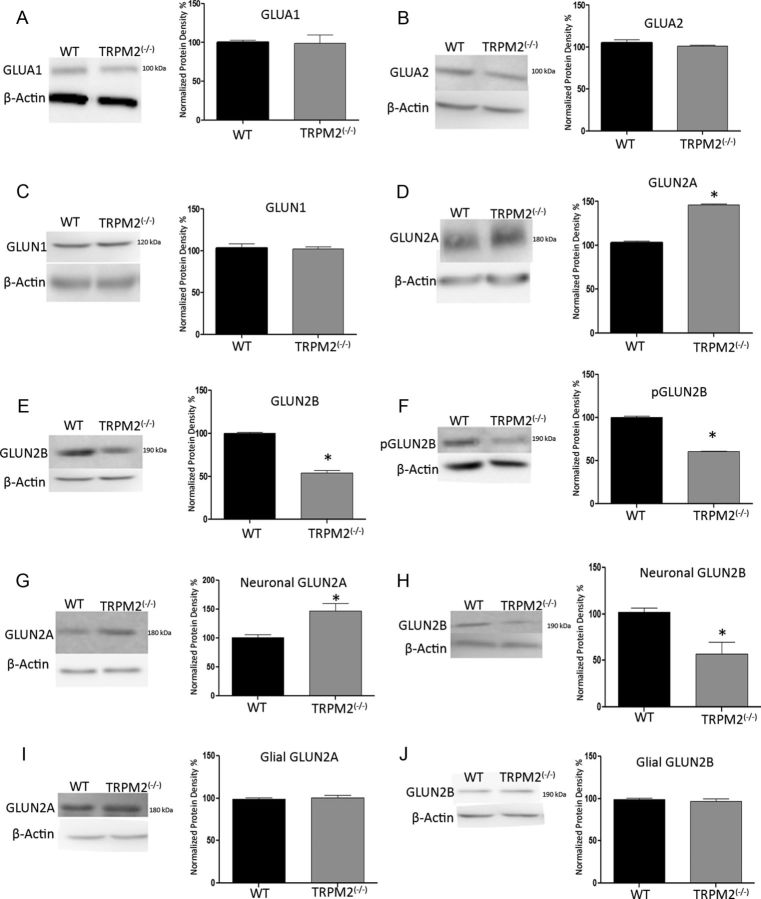
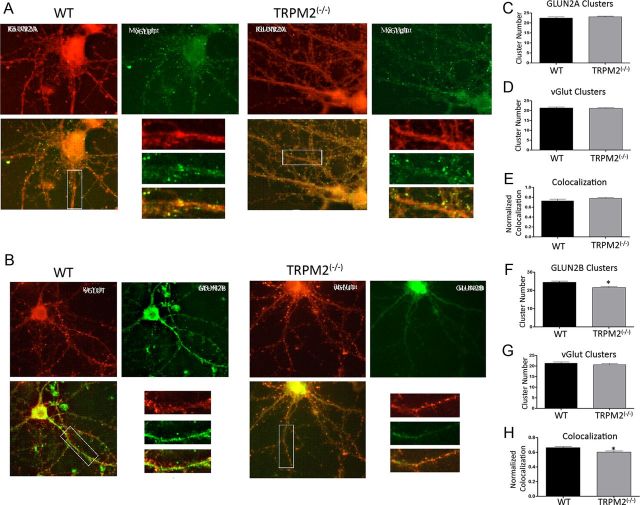
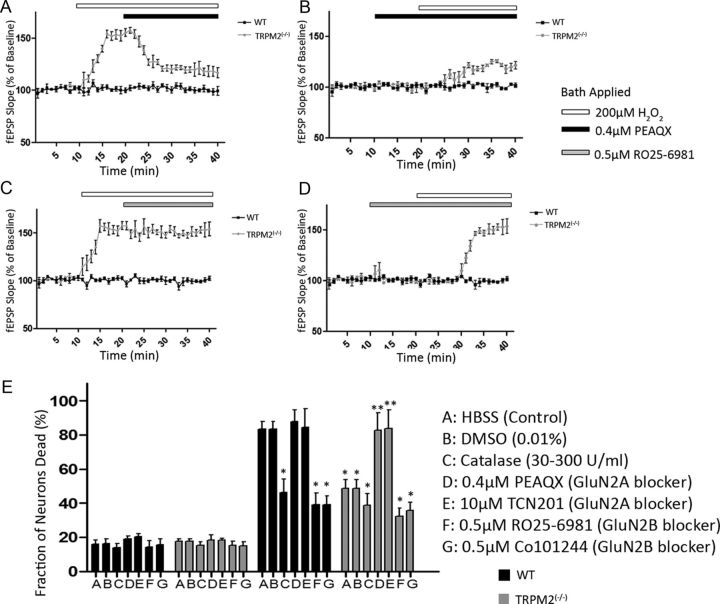
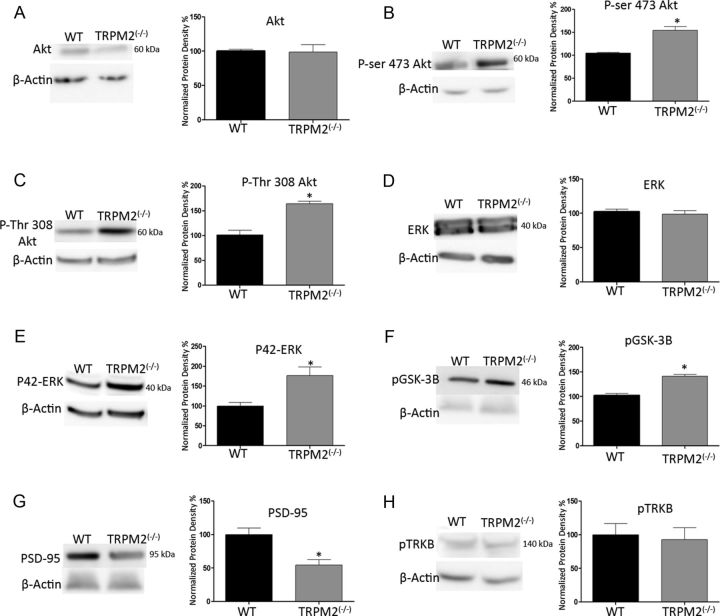
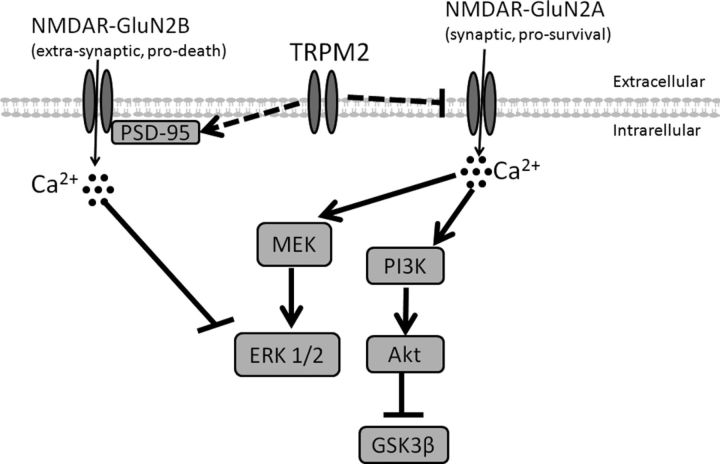
Neuronal vulnerability to ischemia is dependent on the balance between prosurvival and prodeath cellular signaling. In the latter, it is increasingly appreciated that toxic Ca(2+) influx can occur not only via postsynaptic glutamate receptors, but also through other cation conductances. One such conductance, the Transient receptor potential melastatin type-2 (TRPM2) channel, is a nonspecific cation channel having homology to TRPM7, a conductance reported to play a key role in anoxic neuronal death. The role of TRPM2 conductances in ischemic Ca(2+) influx has been difficult to study because of the lack of specific modulators. Here we used TRPM2-null mice (TRPM2(-/-)) to study how TRPM2 may modulate neuronal vulnerability to ischemia. TRPM2(-/-) mice subjected to transient middle cerebral artery occlusion exhibited smaller infarcts when compared with wild-type animals, suggesting that the absence of TRPM2 is neuroprotective. Surprisingly, field potentials (fEPSPs) recorded during redox modulation in brain slices taken from TRPM2(-/-) mice revealed increased excitability, a phenomenon normally associated with ischemic vulnerability, whereas wild-type fEPSPs were unaffected. The upregulation in fEPSP in TRPM2(-/-) neurons was blocked selectively by a GluN2A antagonist. This increase in excitability of TRPM2(-/-) fEPSPs during redox modulation depended on the upregulation and downregulation of GluN2A- and GluN2B-containing NMDARs, respectively, and on augmented prosurvival signaling via Akt and ERK pathways culminating in the inhibition of the proapoptotic factor GSK3β. Our results suggest that TRPM2 plays a role in downregulating prosurvival signals in central neurons and that TRPM2 channels may comprise a therapeutic target for preventing ischemic damage.
Figures
References
-
- Auberson YP, Allgeier H, Bischoff S, Lingenhoehl K, Moretti R, Schmutz M. 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg Med Chem Lett. 2002;12:1099–1102. doi: 10.1016/S0960-894X(02)00074-4. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous