

Modeling precision treatment of breast cancer
- PMID: 24176112
- PMCID: PMC3937590
- DOI: 10.1186/gb-2013-14-10-r110
Modeling precision treatment of breast cancer
Erratum in
-
Erratum to: Modeling precision treatment of breast cancer.Genome Biol. 2015 May 12;16(1):95. doi: 10.1186/s13059-015-0658-5. Genome Biol. 2015. PMID: 25962591 Free PMC article.
Abstract
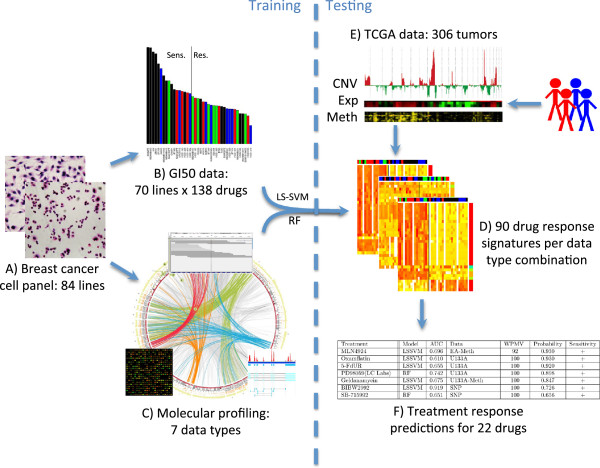
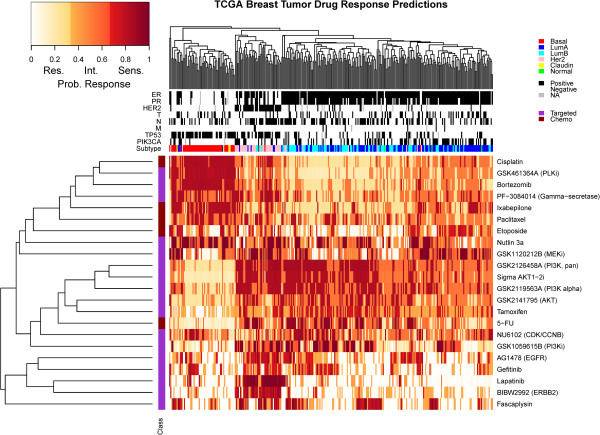
Background: First-generation molecular profiles for human breast cancers have enabled the identification of features that can predict therapeutic response; however, little is known about how the various data types can best be combined to yield optimal predictors. Collections of breast cancer cell lines mirror many aspects of breast cancer molecular pathobiology, and measurements of their omic and biological therapeutic responses are well-suited for development of strategies to identify the most predictive molecular feature sets.
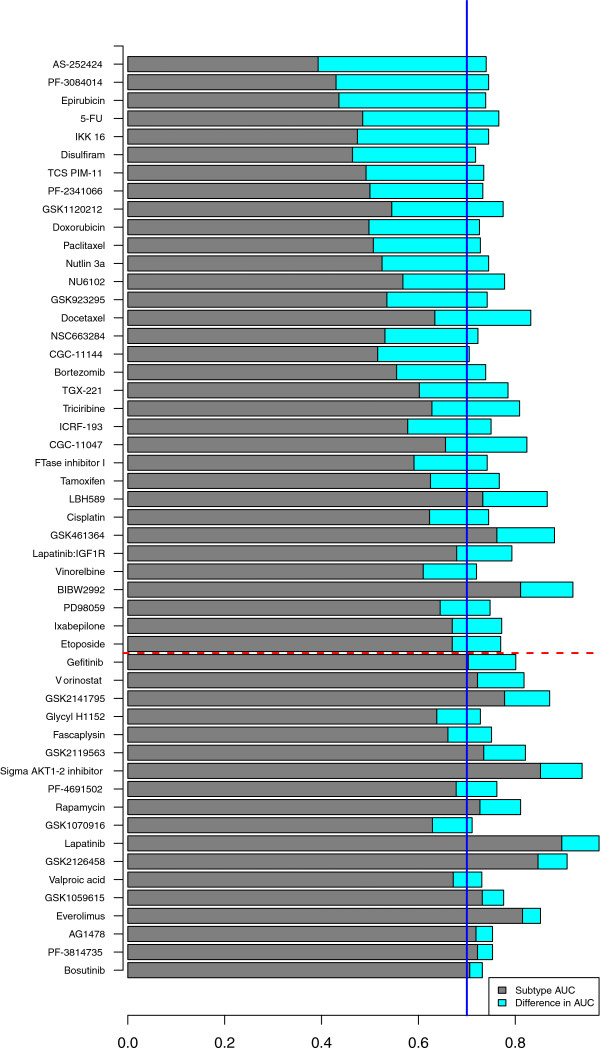
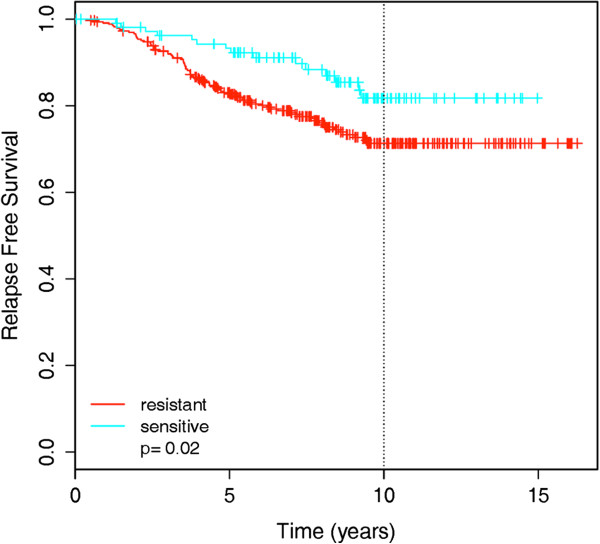
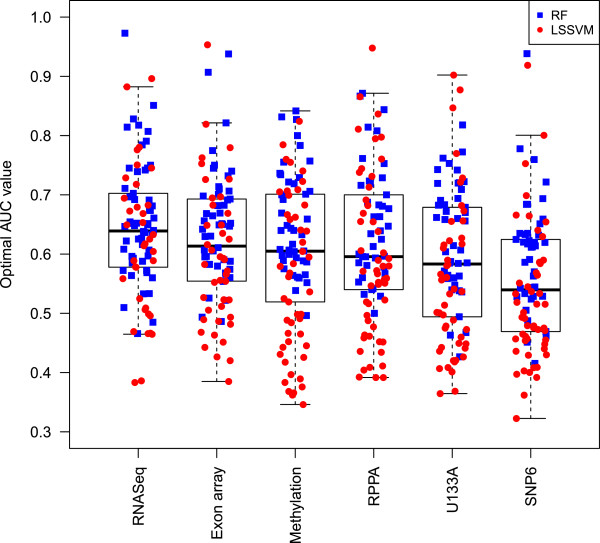
Results: We used least squares-support vector machines and random forest algorithms to identify molecular features associated with responses of a collection of 70 breast cancer cell lines to 90 experimental or approved therapeutic agents. The datasets analyzed included measurements of copy number aberrations, mutations, gene and isoform expression, promoter methylation and protein expression. Transcriptional subtype contributed strongly to response predictors for 25% of compounds, and adding other molecular data types improved prediction for 65%. No single molecular dataset consistently out-performed the others, suggesting that therapeutic response is mediated at multiple levels in the genome. Response predictors were developed and applied to TCGA data, and were found to be present in subsets of those patient samples.
Conclusions: These results suggest that matching patients to treatments based on transcriptional subtype will improve response rates, and inclusion of additional features from other profiling data types may provide additional benefit. Further, we suggest a systems biology strategy for guiding clinical trials so that patient cohorts most likely to respond to new therapies may be more efficiently identified.
Figures
References
-
- Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, Marron JS, Nobel AB, Mardis E, Nielsen TO, Ellis MJ, Perou CM, Bernard PS. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;14:1160–1167. doi: 10.1200/JCO.2008.18.1370. - DOI - PMC - PubMed
-
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Gräf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, Langerød A, Green A, Provenzano E, Wishart G, Pinder S, Watson P, Markowetz F, Murphy L, Ellis I, Purushotham A, Børresen-Dale AL, Brenton JD, Tavaré S. METABRIC Group et al.The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;14:346–352. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
