

EXPANDS: expanding ploidy and allele frequency on nested subpopulations
- PMID: 24177718
- PMCID: PMC3866558
- DOI: 10.1093/bioinformatics/btt622
EXPANDS: expanding ploidy and allele frequency on nested subpopulations
Abstract
Motivation: Several cancer types consist of multiple genetically and phenotypically distinct subpopulations. The underlying mechanism for this intra-tumoral heterogeneity can be explained by the clonal evolution model, whereby growth advantageous mutations cause the expansion of cancer cell subclones. The recurrent phenotype of many cancers may be a consequence of these coexisting subpopulations responding unequally to therapies. Methods to computationally infer tumor evolution and subpopulation diversity are emerging and they hold the promise to improve the understanding of genetic and molecular determinants of recurrence.
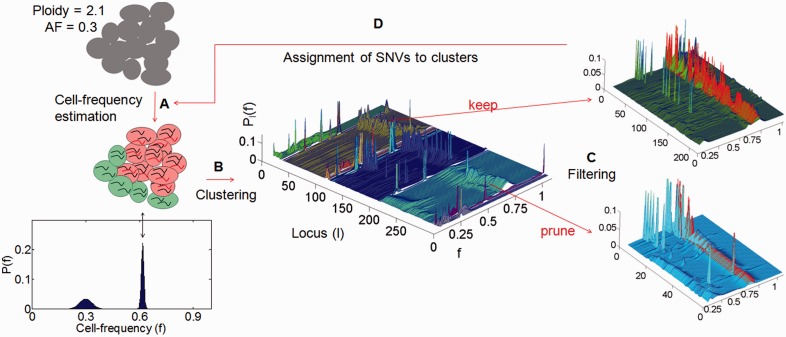
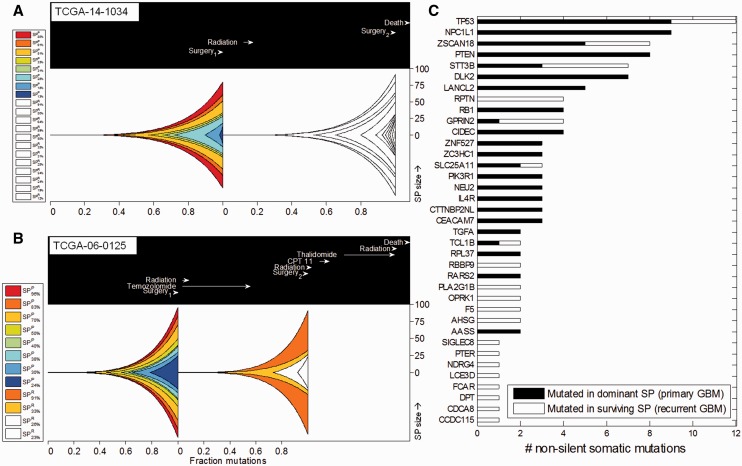
Results: To address cellular subpopulation dynamics within human tumors, we developed a bioinformatic method, EXPANDS. It estimates the proportion of cells harboring specific mutations in a tumor. By modeling cellular frequencies as probability distributions, EXPANDS predicts mutations that accumulate in a cell before its clonal expansion. We assessed the performance of EXPANDS on one whole genome sequenced breast cancer and performed SP analyses on 118 glioblastoma multiforme samples obtained from TCGA. Our results inform about the extent of subclonal diversity in primary glioblastoma, subpopulation dynamics during recurrence and provide a set of candidate genes mutated in the most well-adapted subpopulations. In summary, EXPANDS predicts tumor purity and subclonal composition from sequencing data.
Availability and implementation: EXPANDS is available for download at http://code.google.com/p/expands (matlab version--used in this manuscript) and http://cran.r-project.org/web/packages/expands (R version).
Figures
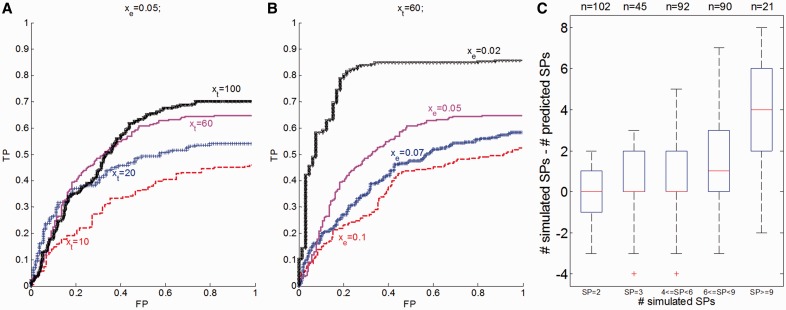
 at a constant noise rate xe = 0.05. (B) A variable noise term
at a constant noise rate xe = 0.05. (B) A variable noise term  was added to the copy number and allele frequency of simulated mutations at a constant number of mutations per clonal expansion xt = 60. (C) Deviation between simulated and predicted number of SPs is shown for various numbers of simulated SPs for all 350 tumors
was added to the copy number and allele frequency of simulated mutations at a constant number of mutations per clonal expansion xt = 60. (C) Deviation between simulated and predicted number of SPs is shown for various numbers of simulated SPs for all 350 tumors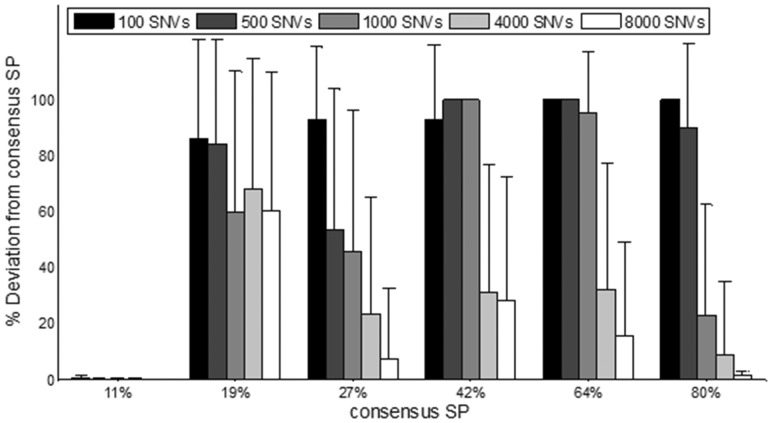
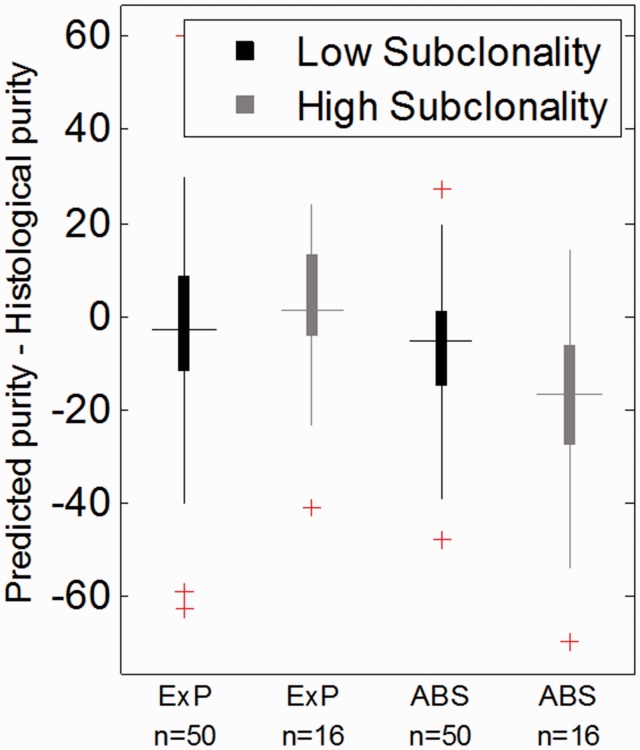
References
-
- Anderson K, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–361. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
