

Pediatric cardiomyopathies: causes, epidemiology, clinical course, preventive strategies and therapies
- PMID: 24180540
- PMCID: PMC3903430
- DOI: 10.2217/fca.13.66
Pediatric cardiomyopathies: causes, epidemiology, clinical course, preventive strategies and therapies
Abstract
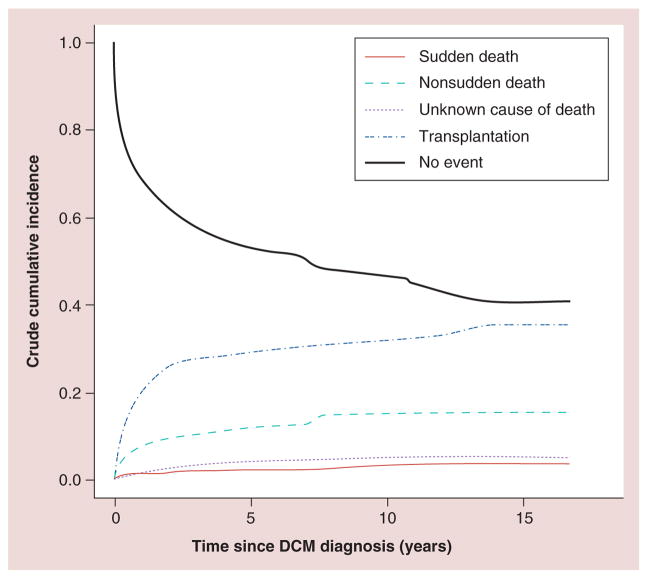
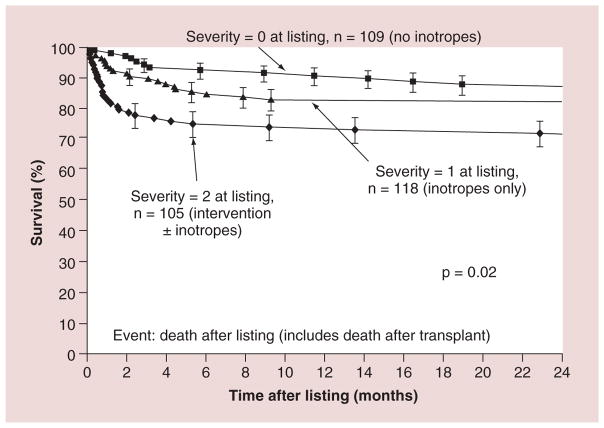
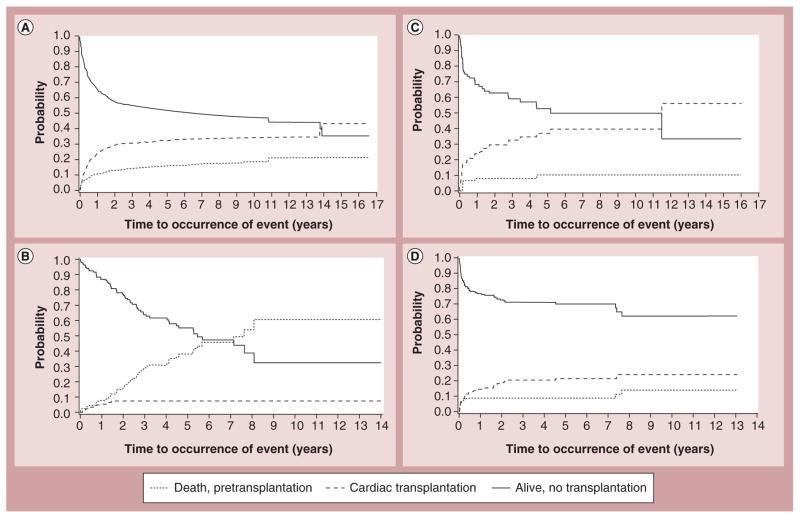
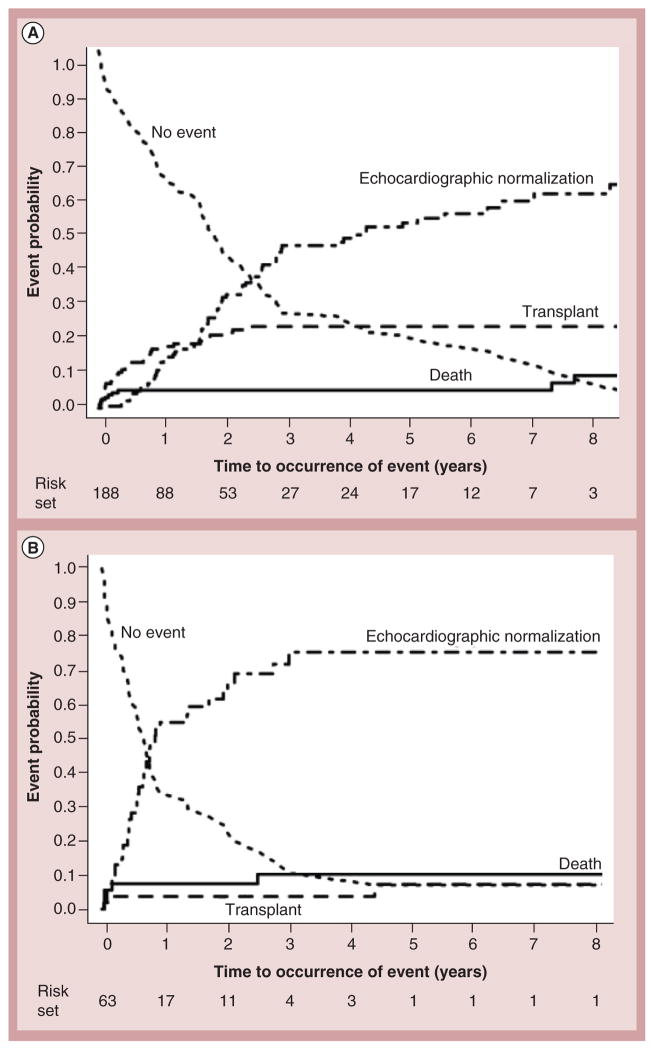
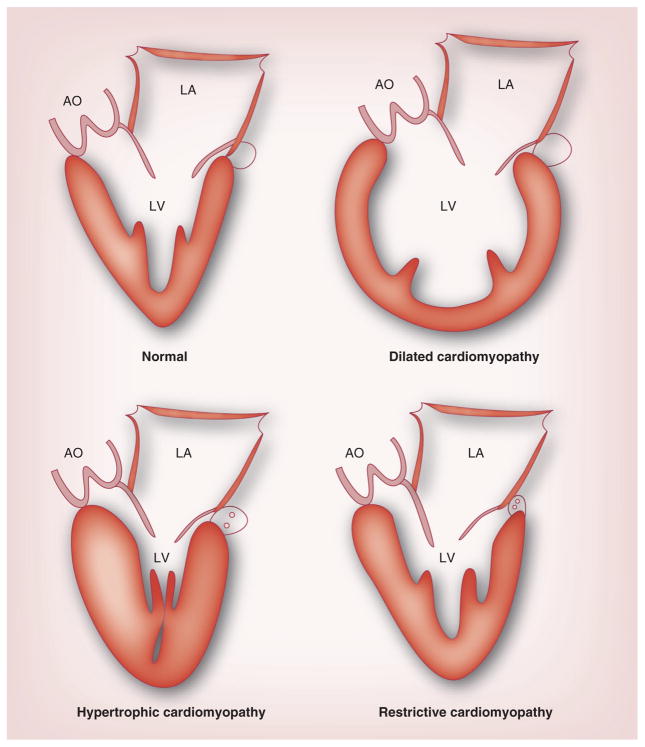
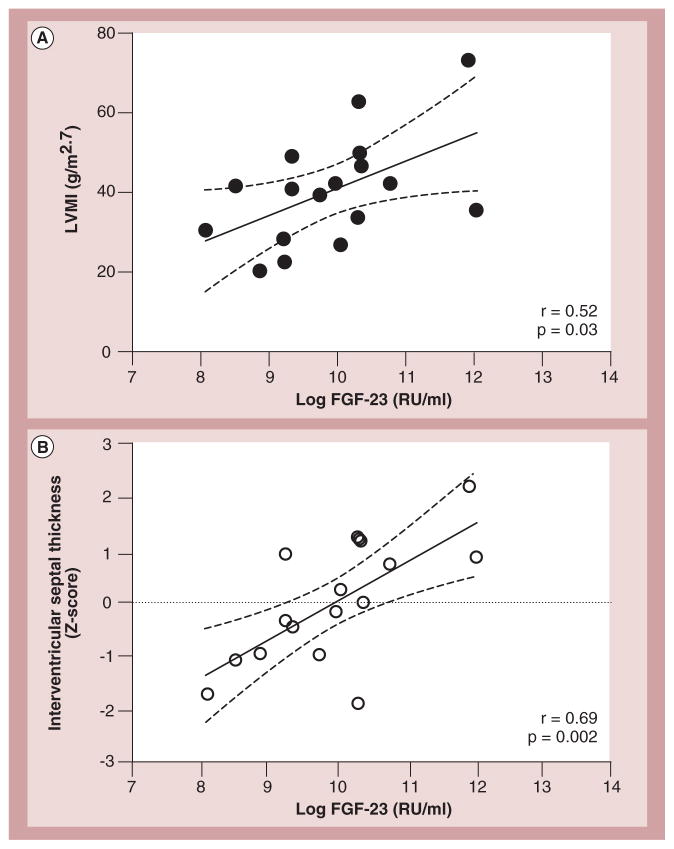
Pediatric cardiomyopathies, which are rare but serious disorders of the muscles of the heart, affect at least one in every 100,000 children in the USA. Approximately 40% of children with symptomatic cardiomyopathy undergo heart transplantation or die from cardiac complications within 2 years. However, a significant number of children suffering from cardiomyopathy are surviving into adulthood, making it an important chronic illness for both pediatric and adult clinicians to understand. The natural history, risk factors, prevalence and incidence of this pediatric condition were not fully understood before the 1990s. Questions regarding optimal diagnostic, prognostic and treatment methods remain. Children require long-term follow-up into adulthood in order to identify the factors associated with best clinical practice including diagnostic approaches, as well as optimal treatment approaches. In this article, we comprehensively review current research on various presentations of this disease, along with current knowledge about their causes, treatments and clinical outcomes.
Figures
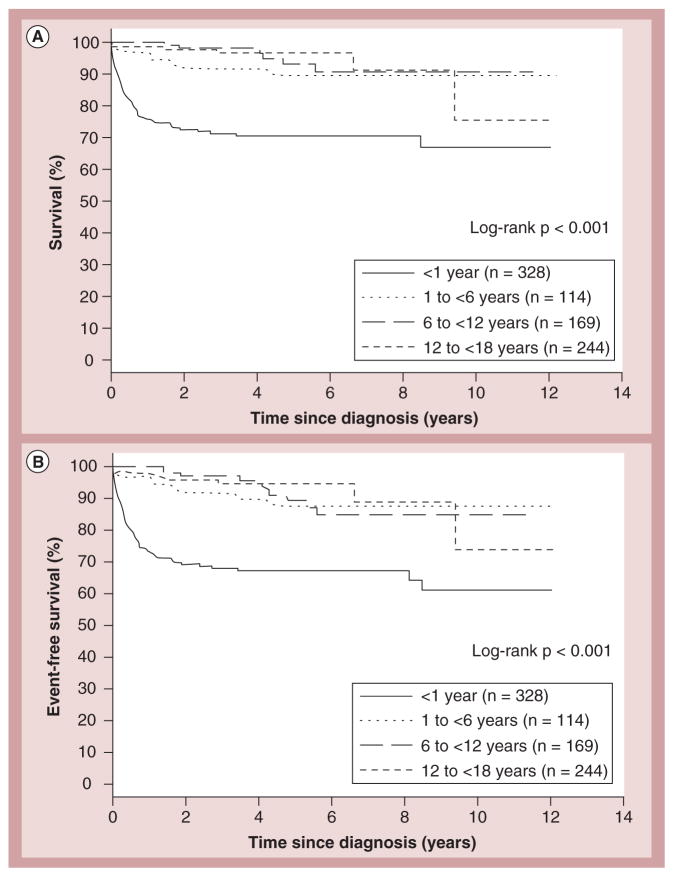
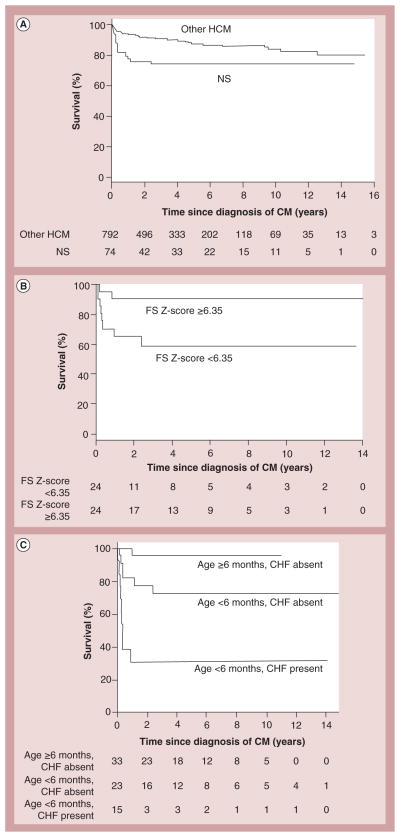
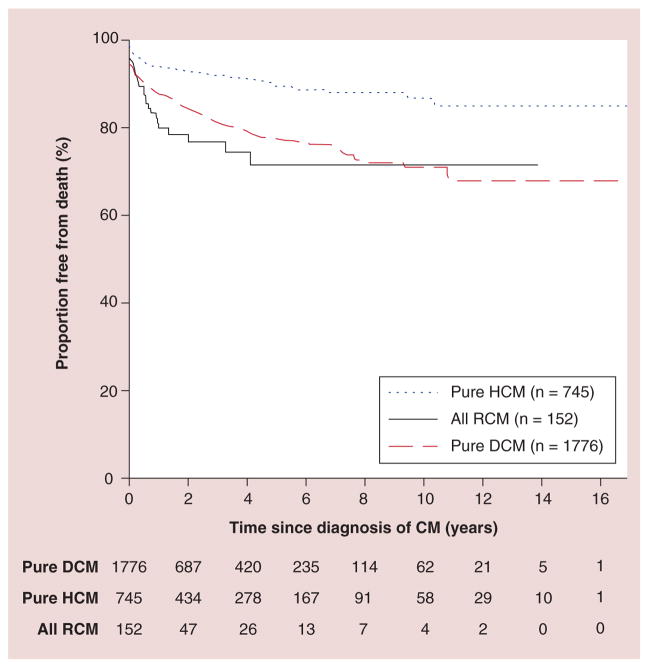
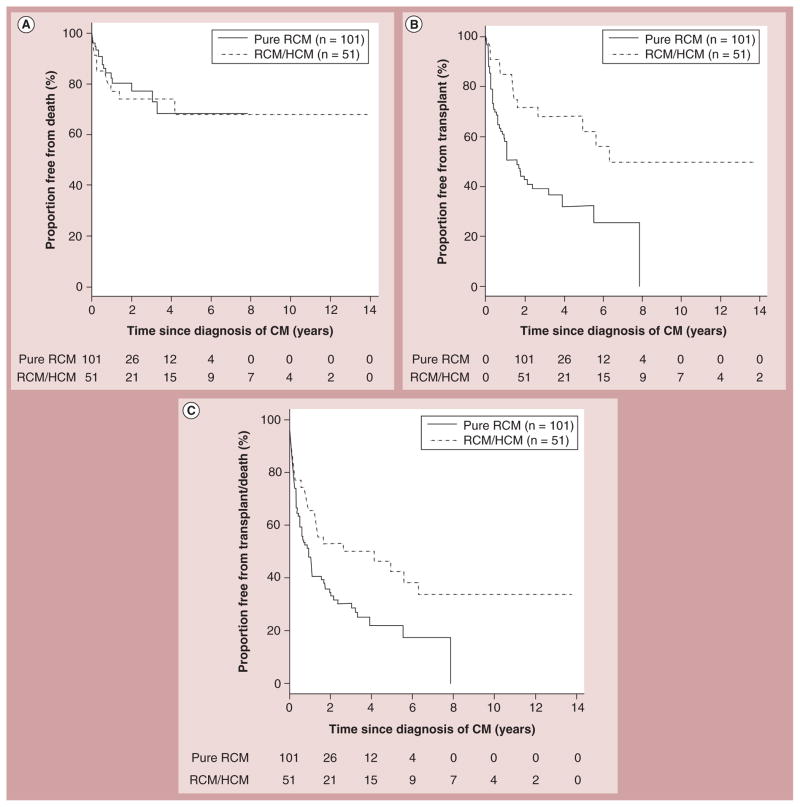
References
-
- Lipshultz SE, Sleeper LA, Towbin JA, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med. 2003;348(17):1647–1655. Found that the incidence of cardiomyopathy was significantly higher in children younger than 1 year old. The estimated incidence of pediatric cardiomyopathy was determined to be 1.13 cases per 100,000 children. Incidence varied according to sex, region and racial origin. - PubMed
-
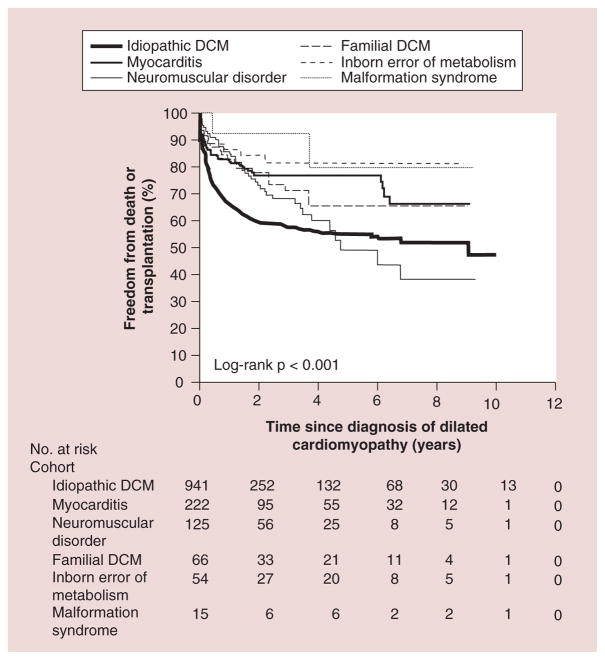
- Towbin JA, Lowe AM, Colan SD, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296(15):1867–1876. Found that the annual incidence of dilated cardiomyopathy in children younger than 18 years of age was 0.57 cases per 100,000. It also found that annual incidence was higher in boys than girls. The majority of children had idiopathic disease, with the most common causes being myocarditis and neuromuscular disease. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical