

Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing
- PMID: 24185094
- PMCID: PMC4180835
- DOI: 10.1038/nbt.2728
Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing
Abstract
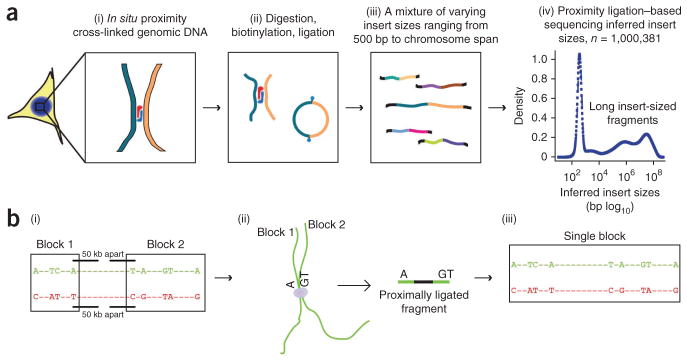
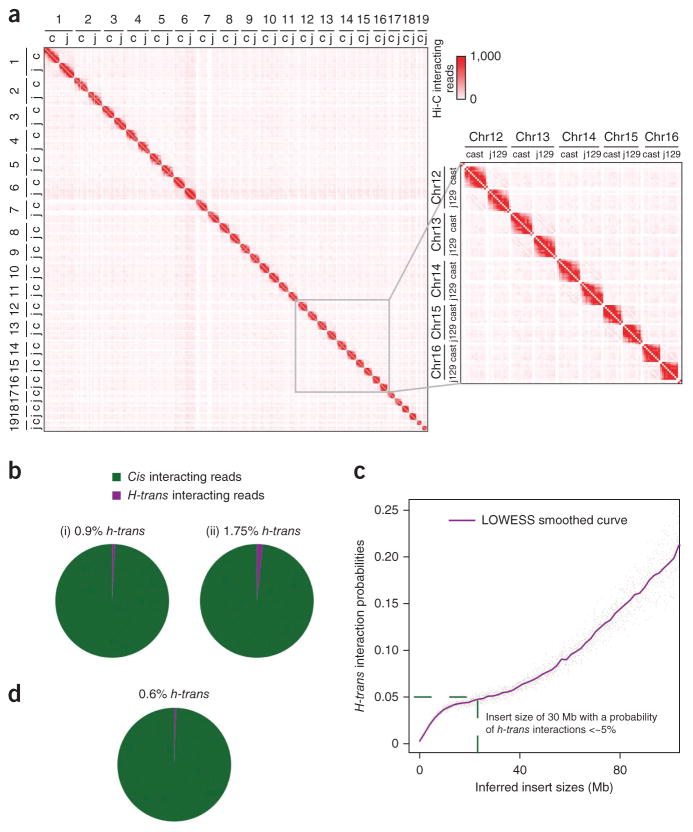
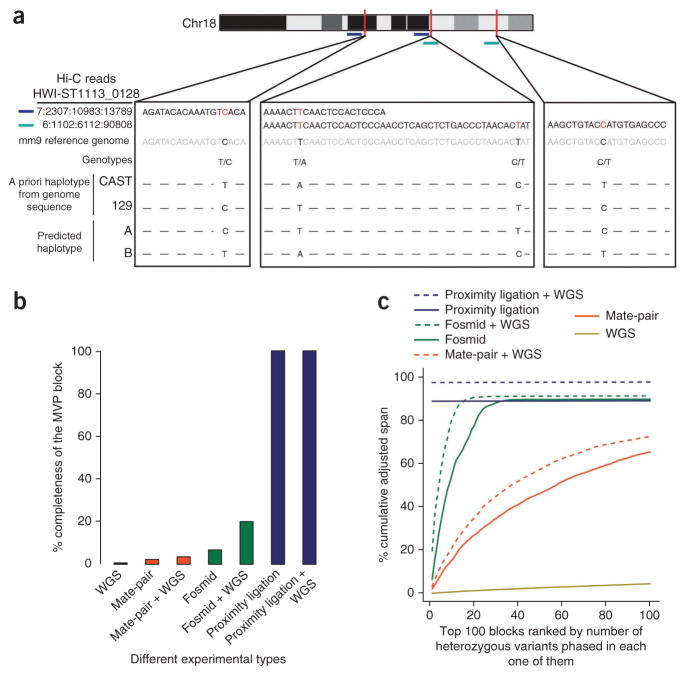
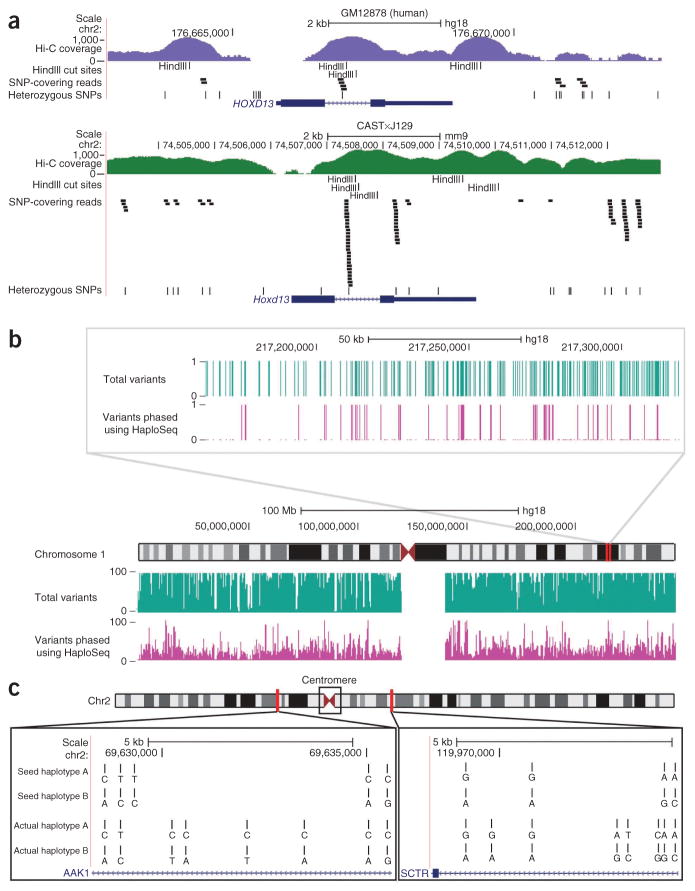
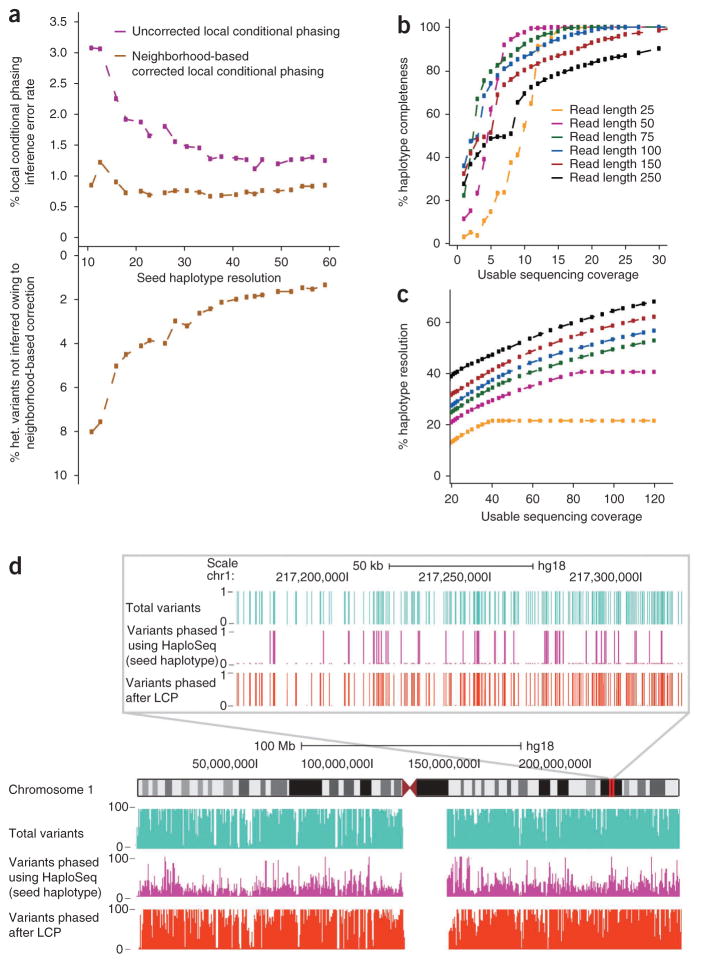
Rapid advances in high-throughput sequencing facilitate variant discovery and genotyping, but linking variants into a single haplotype remains challenging. Here we demonstrate HaploSeq, an approach for assembling chromosome-scale haplotypes by exploiting the existence of 'chromosome territories'. We use proximity ligation and sequencing to show that alleles on homologous chromosomes occupy distinct territories, and therefore this experimental protocol preferentially recovers physically linked DNA variants on a homolog. Computational analysis of such data sets allows for accurate (∼99.5%) reconstruction of chromosome-spanning haplotypes for ∼95% of alleles in hybrid mouse cells with 30× sequencing coverage. To resolve haplotypes for a human genome, which has a low density of variants, we coupled HaploSeq with local conditional phasing to obtain haplotypes for ∼81% of alleles with ∼98% accuracy from just 17× sequencing. Whereas methods based on proximity ligation were originally designed to investigate spatial organization of genomes, our results lend support for their use as a general tool for haplotyping.
Conflict of interest statement
The authors declare competing financial interests: details are available in the online version of the paper.
Figures
Comment in
-
Genome assembly and haplotyping with Hi-C.Nat Biotechnol. 2013 Dec;31(12):1099-101. doi: 10.1038/nbt.2764. Nat Biotechnol. 2013. PMID: 24316648 No abstract available.
References
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
