

Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions
- PMID: 24185095
- PMCID: PMC4117202
- DOI: 10.1038/nbt.2727
Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions
Abstract
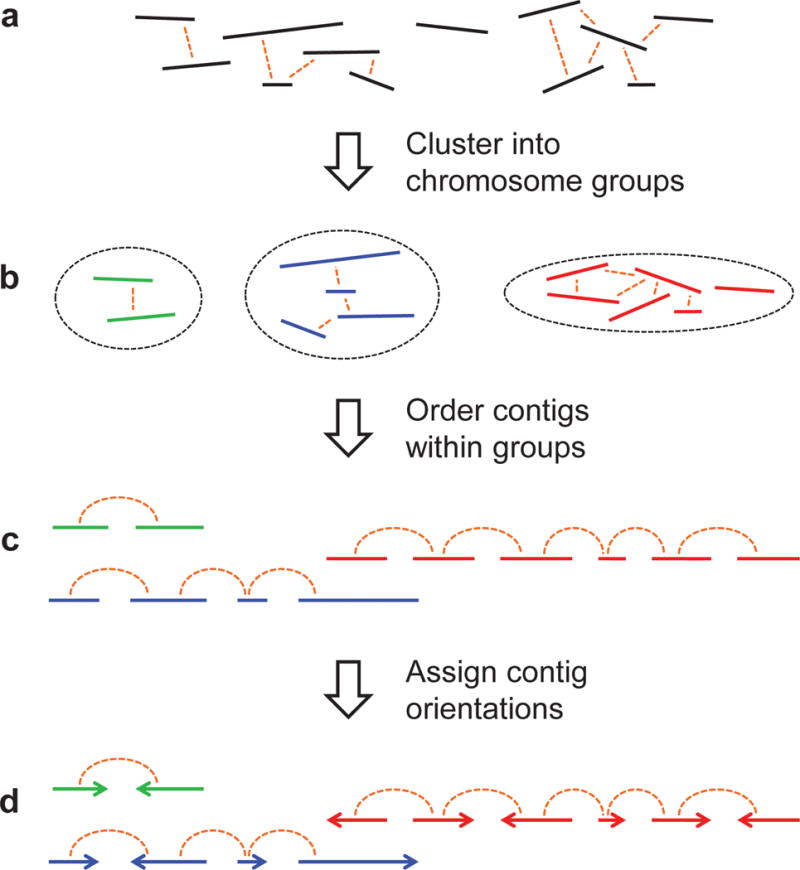
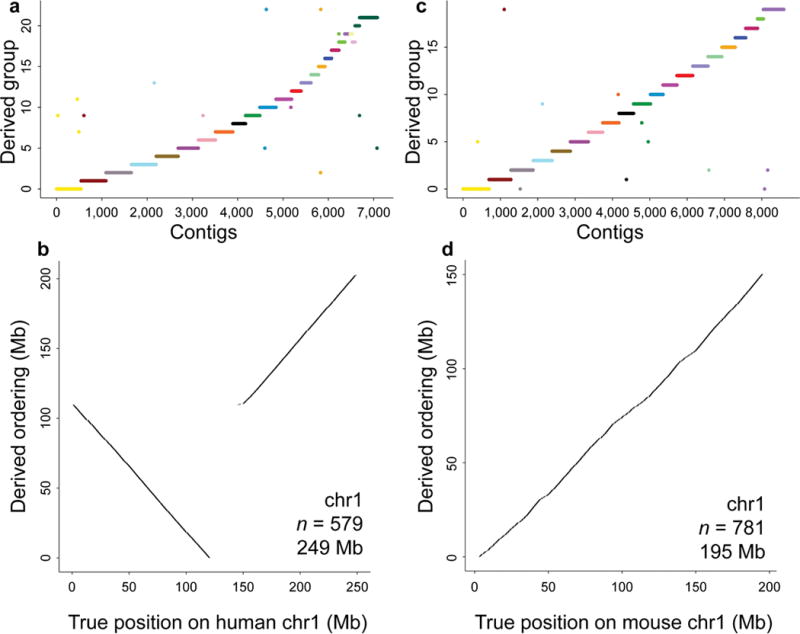
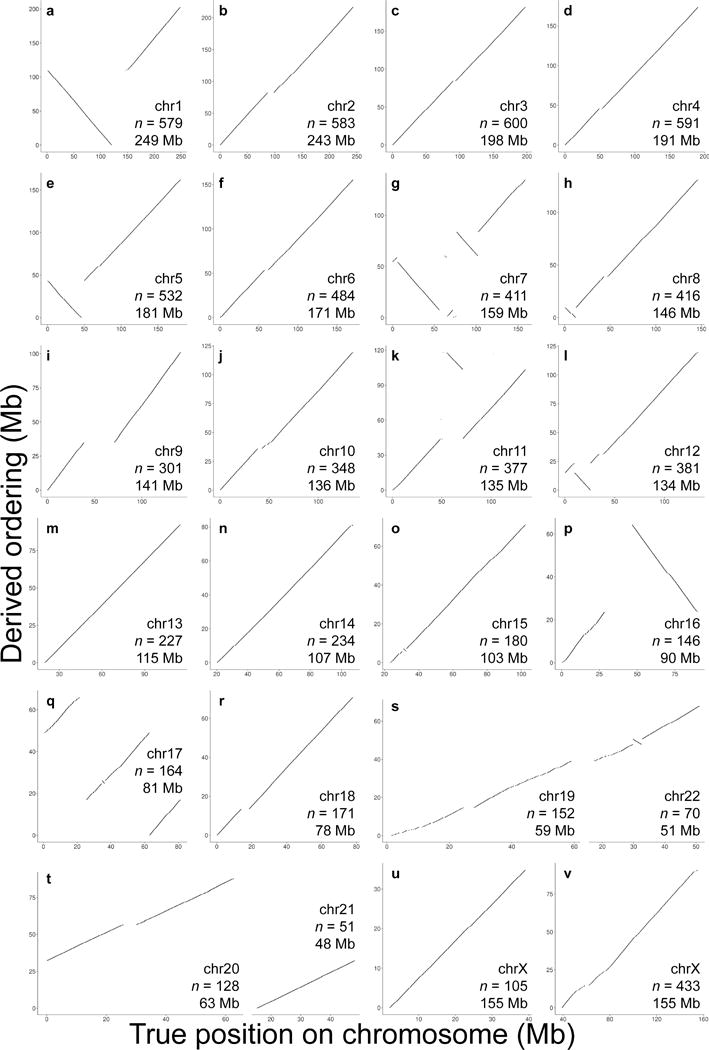
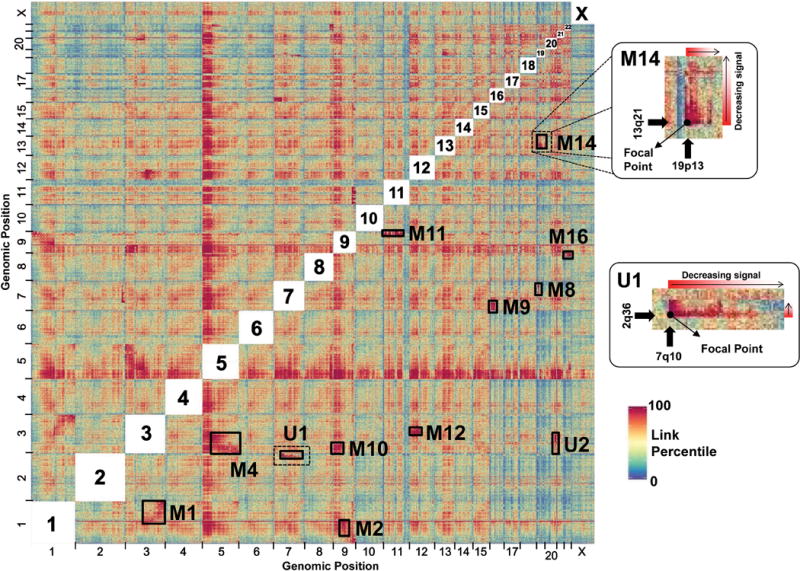
Genomes assembled de novo from short reads are highly fragmented relative to the finished chromosomes of Homo sapiens and key model organisms generated by the Human Genome Project. To address this problem, we need scalable, cost-effective methods to obtain assemblies with chromosome-scale contiguity. Here we show that genome-wide chromatin interaction data sets, such as those generated by Hi-C, are a rich source of long-range information for assigning, ordering and orienting genomic sequences to chromosomes, including across centromeres. To exploit this finding, we developed an algorithm that uses Hi-C data for ultra-long-range scaffolding of de novo genome assemblies. We demonstrate the approach by combining shotgun fragment and short jump mate-pair sequences with Hi-C data to generate chromosome-scale de novo assemblies of the human, mouse and Drosophila genomes, achieving--for the human genome--98% accuracy in assigning scaffolds to chromosome groups and 99% accuracy in ordering and orienting scaffolds within chromosome groups. Hi-C data can also be used to validate chromosomal translocations in cancer genomes.
Conflict of interest statement
The authors are in the process of filing a provisional patent application on this method. J.S. is a member of the scientific advisory board or serves as a consultant for Adaptive Biotechnologies, Ariosa Diagnostics, Stratos Genomics, GenePeeks, Gen9, Good Start Genetics and Rubicon Genomics.
Figures
Comment in
-
Genome assembly and haplotyping with Hi-C.Nat Biotechnol. 2013 Dec;31(12):1099-101. doi: 10.1038/nbt.2764. Nat Biotechnol. 2013. PMID: 24316648 No abstract available.
-
Genomes in 3D improve one-dimensional assemblies.Nat Methods. 2014 Jan;11(1):5. doi: 10.1038/nmeth.2795. Nat Methods. 2014. PMID: 24524125 No abstract available.
References
-
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:1–62.
-
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. - PubMed
-
- Shendure J, Ji H. Next-generation DNA sequencing. Nature Biotechnology. 2008;26:1–11. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
