

Systematic evaluation of spliced alignment programs for RNA-seq data
- PMID: 24185836
- PMCID: PMC4018468
- DOI: 10.1038/nmeth.2722
Systematic evaluation of spliced alignment programs for RNA-seq data
Abstract
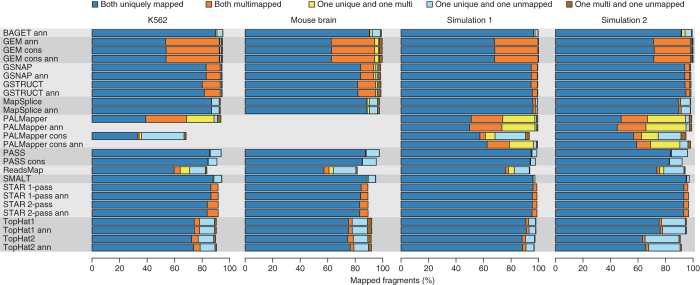
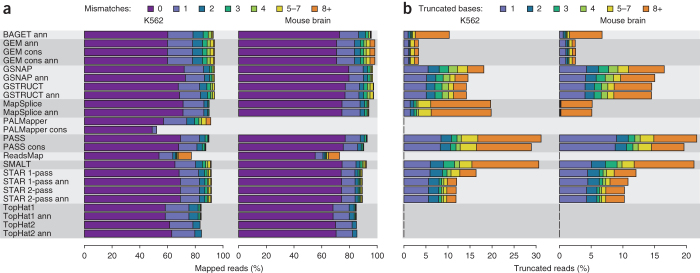
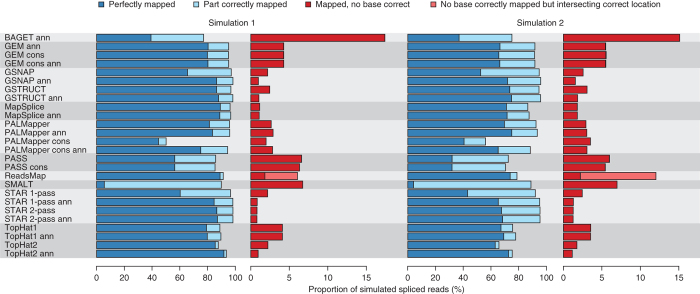
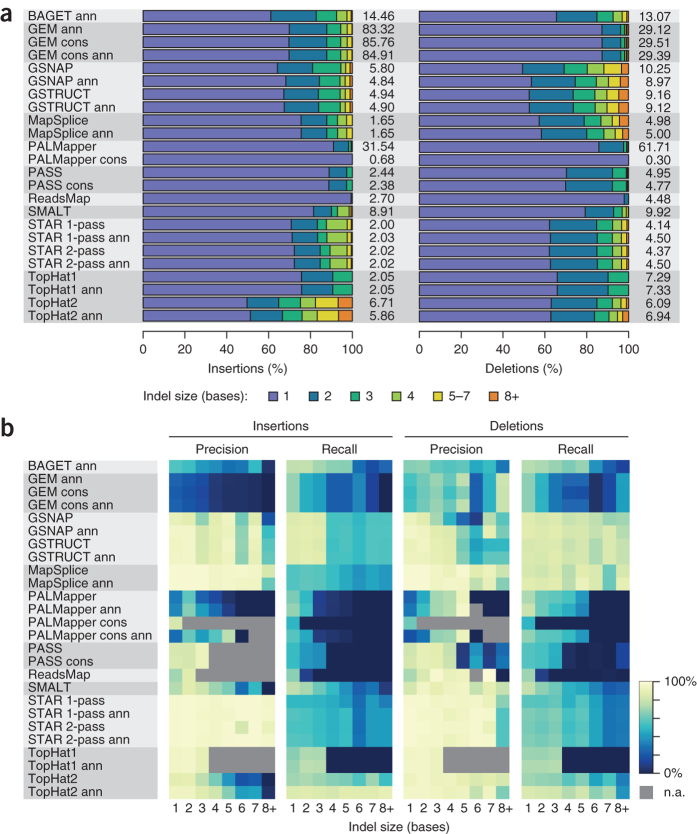
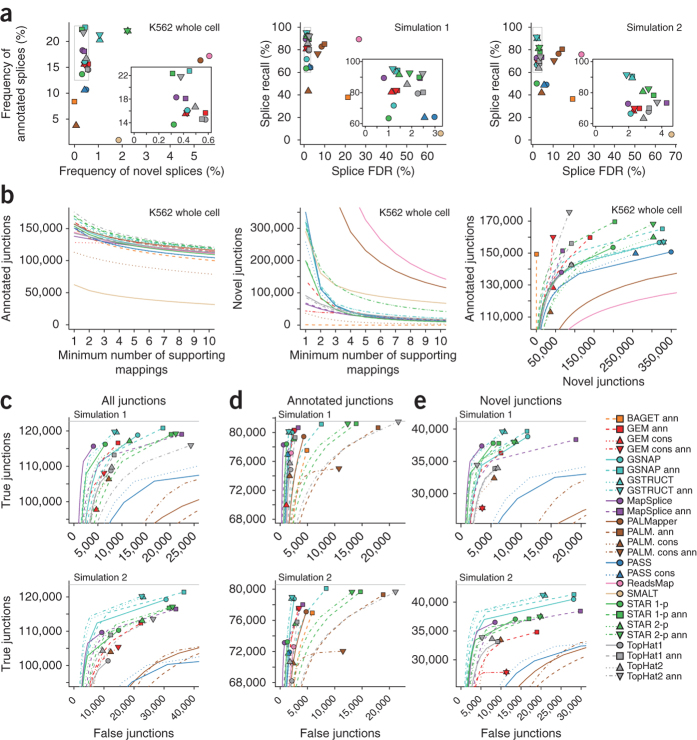
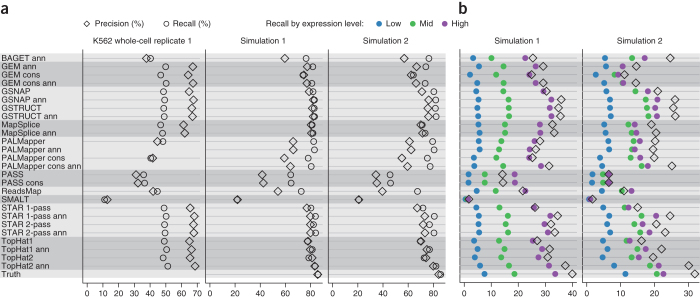
High-throughput RNA sequencing is an increasingly accessible method for studying gene structure and activity on a genome-wide scale. A critical step in RNA-seq data analysis is the alignment of partial transcript reads to a reference genome sequence. To assess the performance of current mapping software, we invited developers of RNA-seq aligners to process four large human and mouse RNA-seq data sets. In total, we compared 26 mapping protocols based on 11 programs and pipelines and found major performance differences between methods on numerous benchmarks, including alignment yield, basewise accuracy, mismatch and gap placement, exon junction discovery and suitability of alignments for transcript reconstruction. We observed concordant results on real and simulated RNA-seq data, confirming the relevance of the metrics employed. Future developments in RNA-seq alignment methods would benefit from improved placement of multimapped reads, balanced utilization of existing gene annotation and a reduced false discovery rate for splice junctions.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Genomics: the state of the art in RNA-seq analysis.Nat Methods. 2013 Dec;10(12):1165-6. doi: 10.1038/nmeth.2735. Nat Methods. 2013. PMID: 24296473 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
