

Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin
- PMID: 24192653
- PMCID: PMC4392818
- DOI: 10.1161/CIRCRESAHA.114.302734
Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin
Abstract
Rationale: Dysfunctional Parkin-mediated mitophagic culling of senescent or damaged mitochondria is a major pathological process underlying Parkinson disease and a potential genetic mechanism of cardiomyopathy. Despite epidemiological associations between Parkinson disease and heart failure, the role of Parkin and mitophagic quality control in maintaining normal cardiac homeostasis is poorly understood.
Objective: We used germline mutants and cardiac-specific RNA interference to interrogate Parkin regulation of cardiomyocyte mitochondria and examine functional crosstalk between mitophagy and mitochondrial dynamics in Drosophila heart tubes.
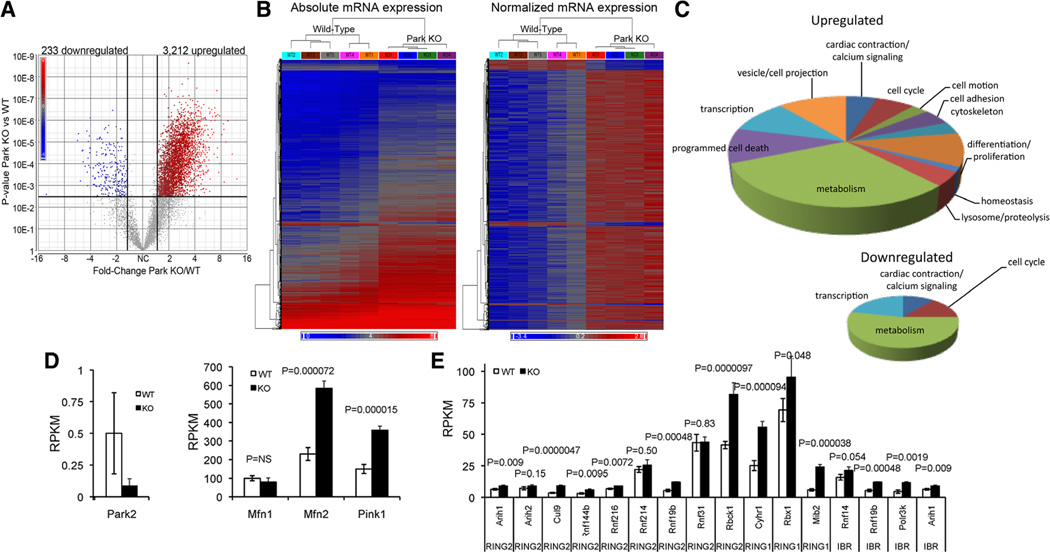
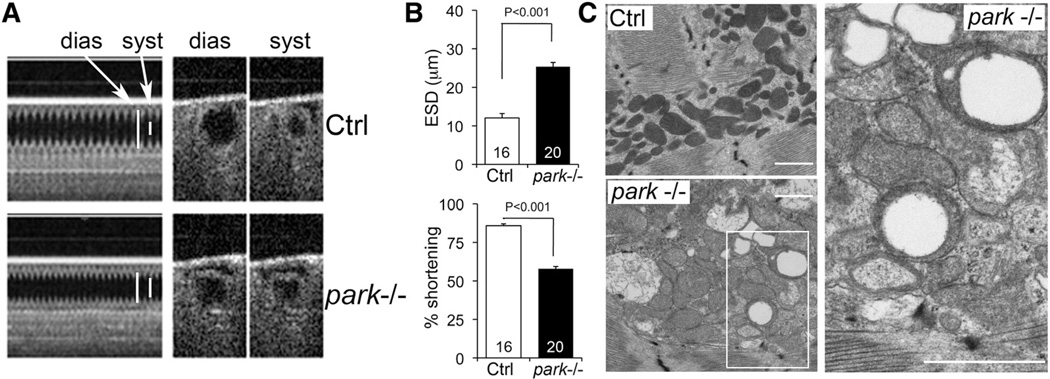
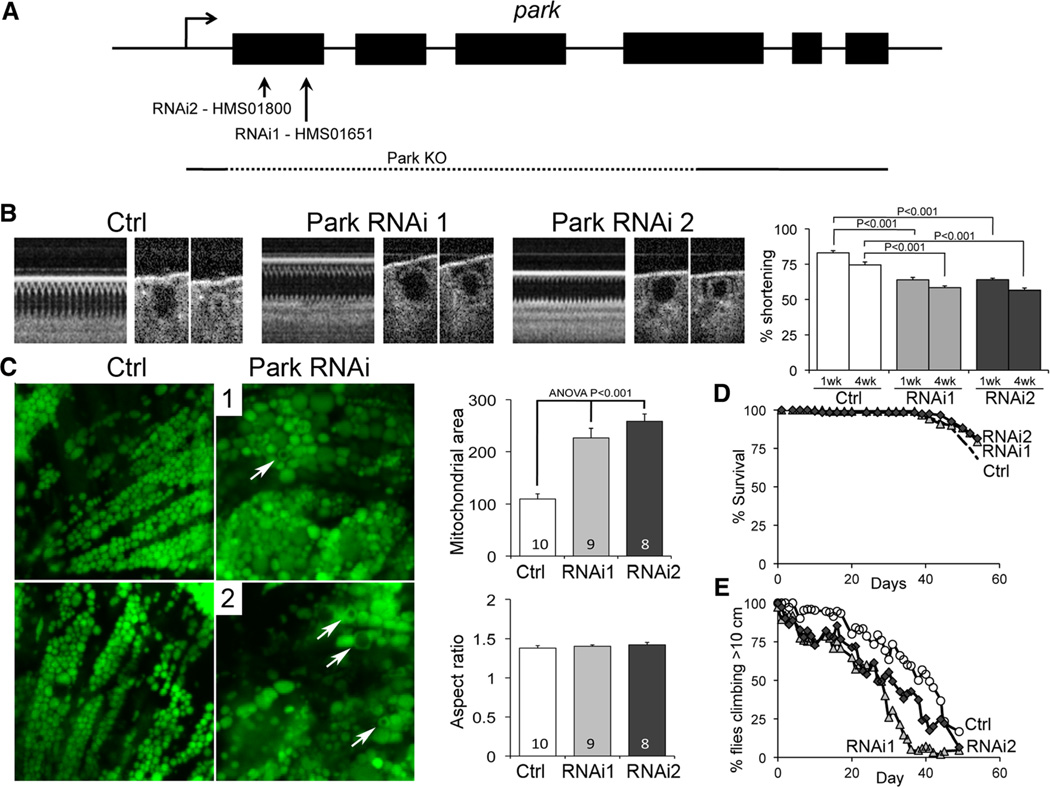
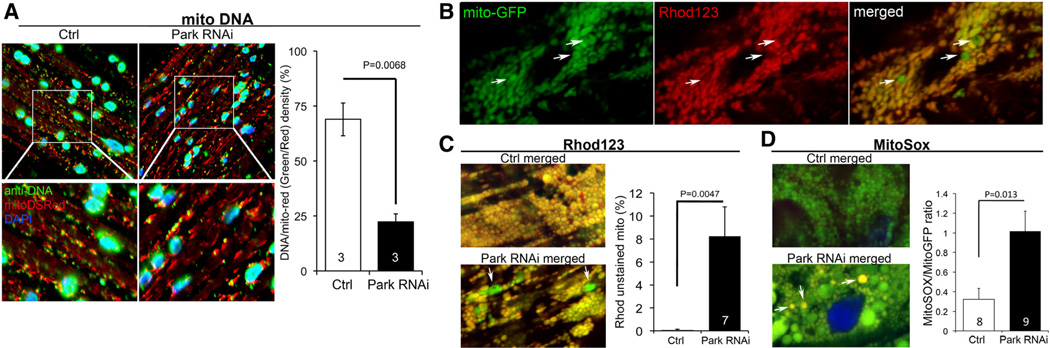
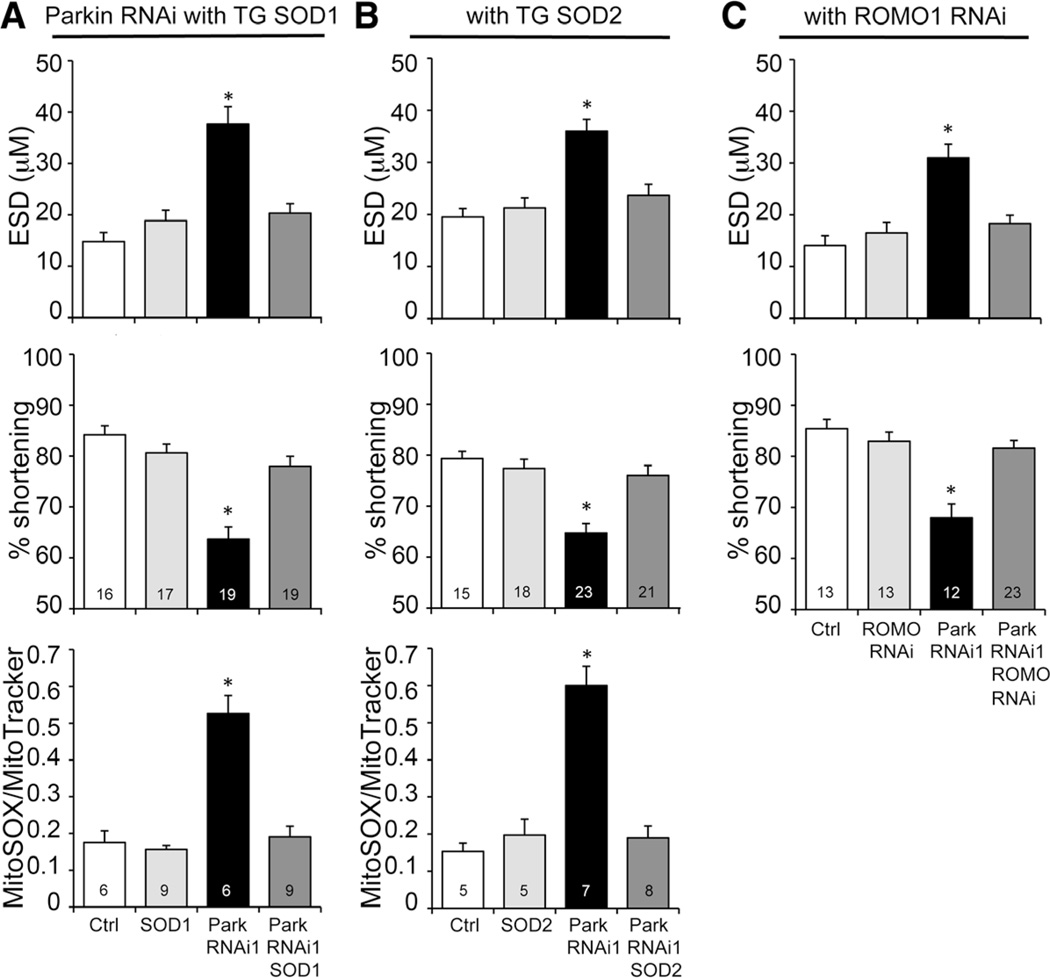
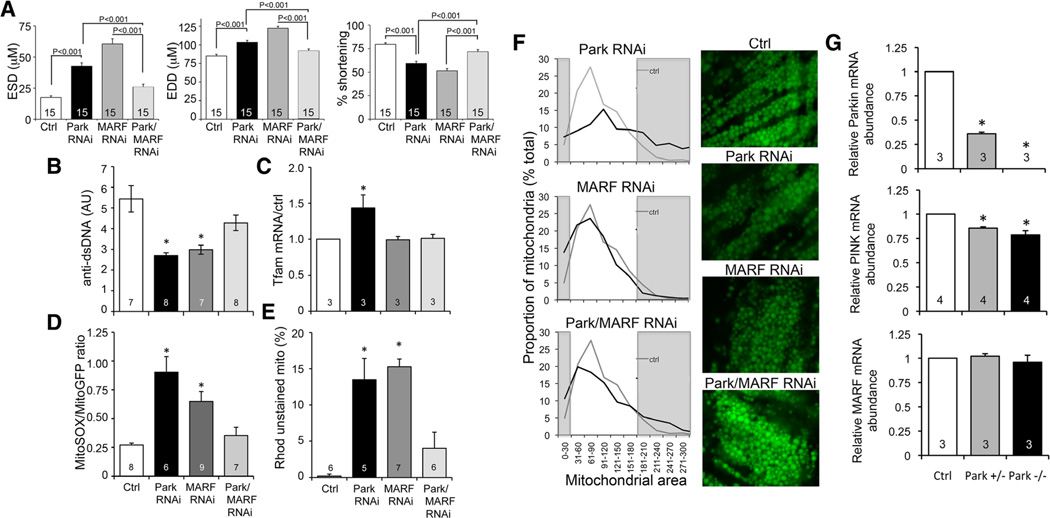
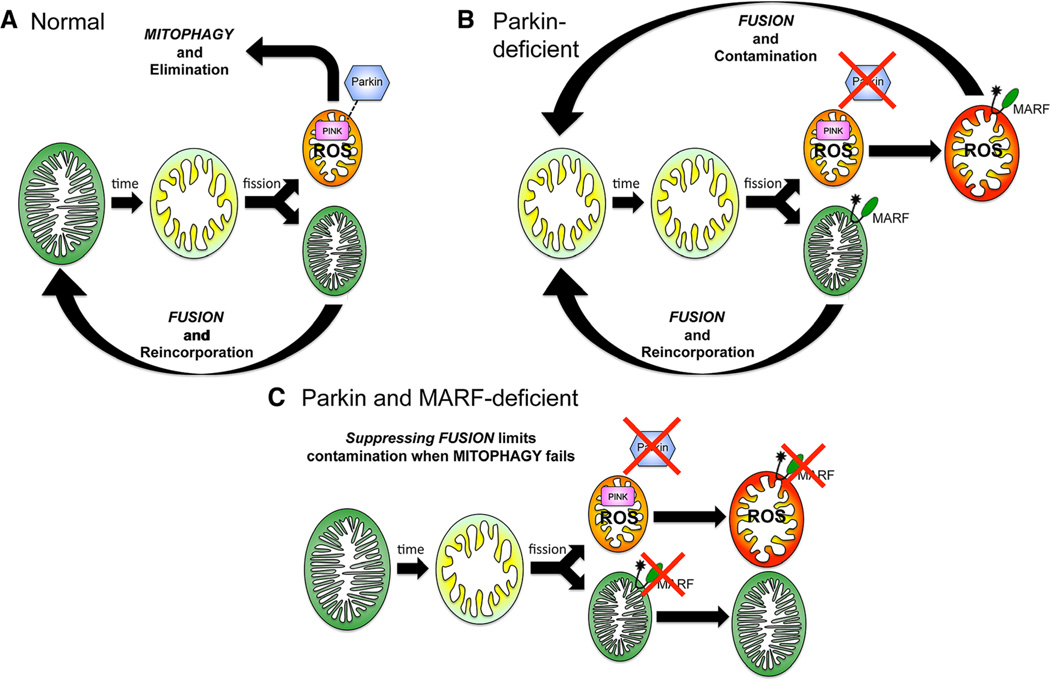
Methods and results: Transcriptional profiling of Parkin knockout mouse hearts revealed compensatory upregulation of multiple related E3 ubiquitin ligases. Because Drosophila lack most of these redundant genes, we examined heart tubes of parkin knockout flies and observed accumulation of enlarged hollow donut mitochondria with dilated cardiomyopathy, which could be rescued by cardiomyocyte-specific Parkin expression. Identical abnormalities were induced by cardiomyocyte-specific Parkin suppression using 2 different inhibitory RNAs. Parkin-deficient cardiomyocyte mitochondria exhibited dysmorphology, depolarization, and reactive oxygen species generation without calcium cycling abnormalities, pointing to a primary mitochondrial defect. Suppressing cardiomyocyte mitochondrial fusion in Parkin-deficient fly heart tubes completely prevented the cardiomyopathy and corrected mitochondrial dysfunction without normalizing mitochondrial dysmorphology, demonstrating a central role for mitochondrial fusion in the cardiomyopathy provoked by impaired mitophagy.
Conclusions: Parkin deficiency and resulting mitophagic disruption produces cardiomyopathy in part by contamination of the cardiomyocyte mitochondrial pool through fusion between improperly retained dysfunctional/senescent and normal mitochondria. Limiting mitochondrial contagion by inhibiting organelle fusion shows promise for minimizing organ dysfunction produced by defective mitophagic signaling.
Keywords: cardiomyopathies; mitochondrial degradation; mitochondrial dynamics.
Figures
References
-
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. - PubMed
-
- Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
