

doi: 10.1534/g3.113.008797.
Performance of high-throughput sequencing for the discovery of genetic variation across the complete size spectrum
Affiliations
- PMID: 24192839
- PMCID: PMC3887540
- DOI: 10.1534/g3.113.008797
Item in Clipboard
Performance of high-throughput sequencing for the discovery of genetic variation across the complete size spectrum
G3 (Bethesda).
.
Abstract
We observed that current high-throughput sequencing approaches only detected a fraction of the full size-spectrum of insertions, deletions, and copy number variants compared with a previously published, Sanger-sequenced human genome. The sensitivity for detection was the lowest in the 100- to 10,000-bp size range, and at DNA repeats, with copy number gains harder to delineate than losses. We discuss strategies for discovering the full spectrum of genetic variation necessary for disease association studies.
Keywords: copy number variation; genome variation annotation; high-throughput sequencing; insertion/deletion.
Figures
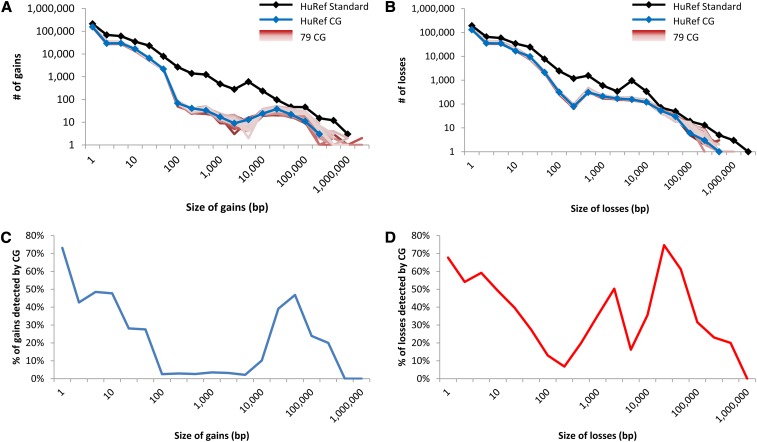
Variation distribution of genomes sequenced. The size distribution of nonredundant (A) gains and (B) losses detected in the HuRef and 79 other samples. The proportion of nonredundant (C) gains and (D) losses detected in HuRef by CG in comparison with HuRef Standard.
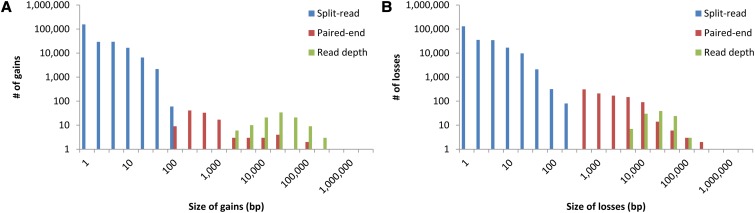
Size distribution of HuRef CG gains and losses detected by each discovery strategy examined: split-read, paired-end mapping and read depth. (A) Gains. (B) Losses.
References
-
- Drmanac R., Sparks A. B., Callow M. J., Halpern A. L., Burns N. L., et al. , 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327: 78–81. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources