

Posterior predictive Bayesian phylogenetic model selection
- PMID: 24193892
- PMCID: PMC3985471
- DOI: 10.1093/sysbio/syt068
Posterior predictive Bayesian phylogenetic model selection
Abstract
We present two distinctly different posterior predictive approaches to Bayesian phylogenetic model selection and illustrate these methods using examples from green algal protein-coding cpDNA sequences and flowering plant rDNA sequences. The Gelfand-Ghosh (GG) approach allows dissection of an overall measure of model fit into components due to posterior predictive variance (GGp) and goodness-of-fit (GGg), which distinguishes this method from the posterior predictive P-value approach. The conditional predictive ordinate (CPO) method provides a site-specific measure of model fit useful for exploratory analyses and can be combined over sites yielding the log pseudomarginal likelihood (LPML) which is useful as an overall measure of model fit. CPO provides a useful cross-validation approach that is computationally efficient, requiring only a sample from the posterior distribution (no additional simulation is required). Both GG and CPO add new perspectives to Bayesian phylogenetic model selection based on the predictive abilities of models and complement the perspective provided by the marginal likelihood (including Bayes Factor comparisons) based solely on the fit of competing models to observed data.
Figures
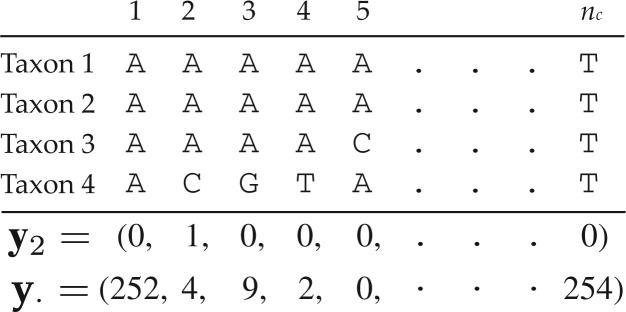
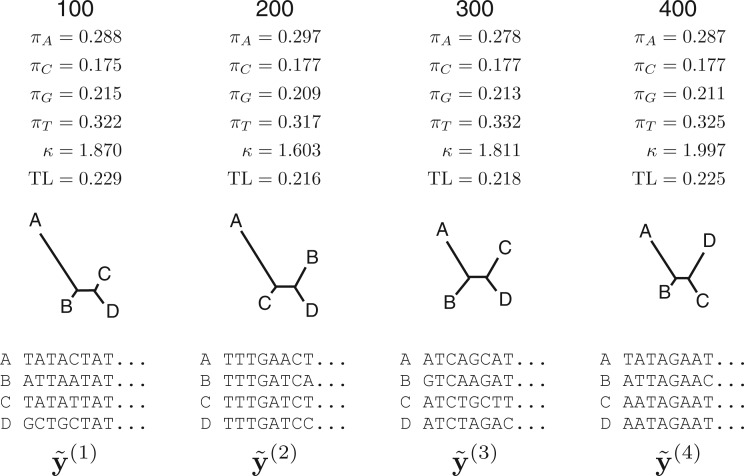
 under a HKY substitution model for a four-taxon problem. Numbers at the top represent the MCMC iteration at which a posterior sample was drawn, TL=tree length (sum of the five edge length parameters), κ =transition/transversion rate ratio, and πA, πC, πG, πT =nucleotide equilibrium relative frequencies. For each iteration, the sampled parameter values and tree are used to simulate one data set, the vector of pattern counts of which constitute
under a HKY substitution model for a four-taxon problem. Numbers at the top represent the MCMC iteration at which a posterior sample was drawn, TL=tree length (sum of the five edge length parameters), κ =transition/transversion rate ratio, and πA, πC, πG, πT =nucleotide equilibrium relative frequencies. For each iteration, the sampled parameter values and tree are used to simulate one data set, the vector of pattern counts of which constitute  .
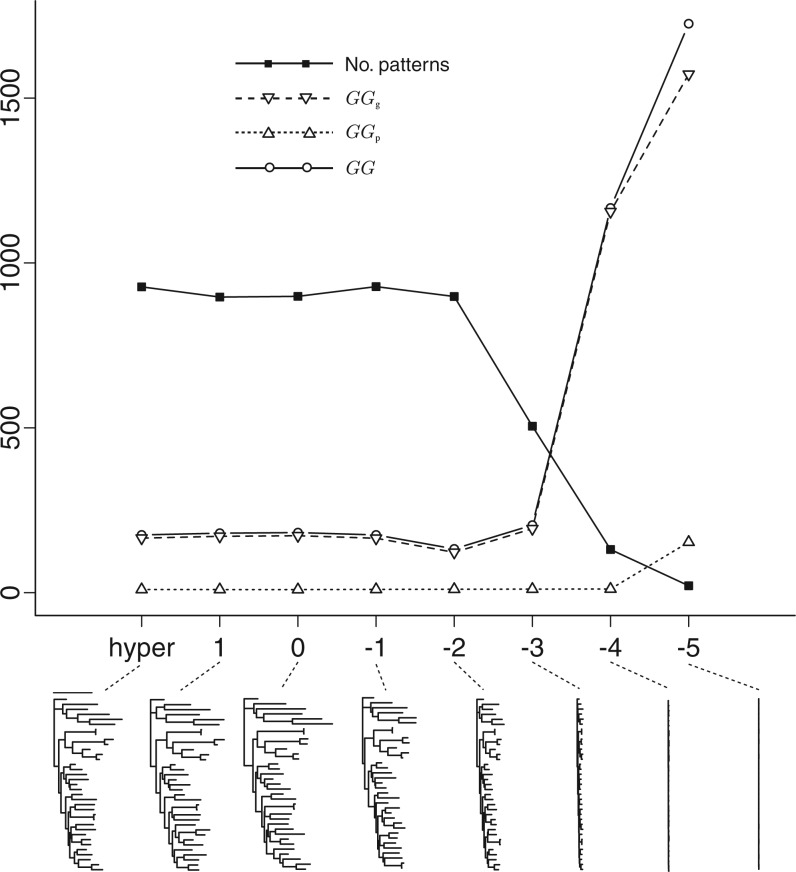
.
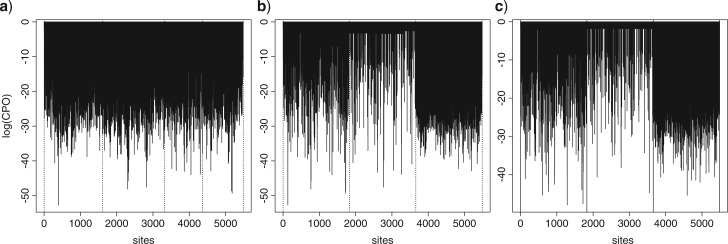
Similar articles
-
Phycas: software for Bayesian phylogenetic analysis.Syst Biol. 2015 May;64(3):525-31. doi: 10.1093/sysbio/syu132. Epub 2015 Jan 9. Syst Biol. 2015. PMID: 25577605
-
Data partitions and complex models in Bayesian analysis: the phylogeny of Gymnophthalmid lizards.Syst Biol. 2004 Jun;53(3):448-69. doi: 10.1080/10635150490445797. Syst Biol. 2004. PMID: 15503673
-
Marginal Likelihoods in Phylogenetics: A Review of Methods and Applications.Syst Biol. 2019 Sep 1;68(5):681-697. doi: 10.1093/sysbio/syz003. Syst Biol. 2019. PMID: 30668834 Free PMC article. Review.
-
How Well Does Your Phylogenetic Model Fit Your Data?Syst Biol. 2019 Jan 1;68(1):157-167. doi: 10.1093/sysbio/syy066. Syst Biol. 2019. PMID: 30329125
-
Modeling compositional heterogeneity.Syst Biol. 2004 Jun;53(3):485-95. doi: 10.1080/10635150490445779. Syst Biol. 2004. PMID: 15503675
Cited by
-
Identifying model violations under the multispecies coalescent model using P2C2M.SNAPP.PeerJ. 2020 Jan 10;8:e8271. doi: 10.7717/peerj.8271. eCollection 2020. PeerJ. 2020. PMID: 31949994 Free PMC article.
-
Differences in Performance among Test Statistics for Assessing Phylogenomic Model Adequacy.Genome Biol Evol. 2018 Jun 1;10(6):1375-1388. doi: 10.1093/gbe/evy094. Genome Biol Evol. 2018. PMID: 29788113 Free PMC article.
-
Bayesian Total-Evidence Dating Reveals the Recent Crown Radiation of Penguins.Syst Biol. 2017 Jan 1;66(1):57-73. doi: 10.1093/sysbio/syw060. Syst Biol. 2017. PMID: 28173531 Free PMC article.
-
Chloroplast Phylogenomic Inference of Green Algae Relationships.Sci Rep. 2016 Feb 5;6:20528. doi: 10.1038/srep20528. Sci Rep. 2016. PMID: 26846729 Free PMC article.
-
Assessing model adequacy for Bayesian Skyline plots using posterior predictive simulation.PLoS One. 2022 Jul 25;17(7):e0269438. doi: 10.1371/journal.pone.0269438. eCollection 2022. PLoS One. 2022. PMID: 35877611 Free PMC article.
References
-
- Akaike H. A new look at statistical model identification. IEEE Trans. Automat. Contr. 1974;19:716–723.
-
- Arima S., Tardella L. Improved harmonic mean estimator for phylogenetic model evidence. J. Comput. Biol. 2012;19:418–438. - PubMed
-
- Bergthorsson U., Adams K.L., Thomason B., Palmer J.D. Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature. 2003;424:197–201. - PubMed
-
- Bollback J. Bayesian model adequacy and choice in phylogenetics. Mol. Biol. Evol. 2002;19:1171–1180. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
