

VDR attenuates acute lung injury by blocking Ang-2-Tie-2 pathway and renin-angiotensin system
- PMID: 24196349
- PMCID: PMC3857197
- DOI: 10.1210/me.2013-1146
VDR attenuates acute lung injury by blocking Ang-2-Tie-2 pathway and renin-angiotensin system
Abstract
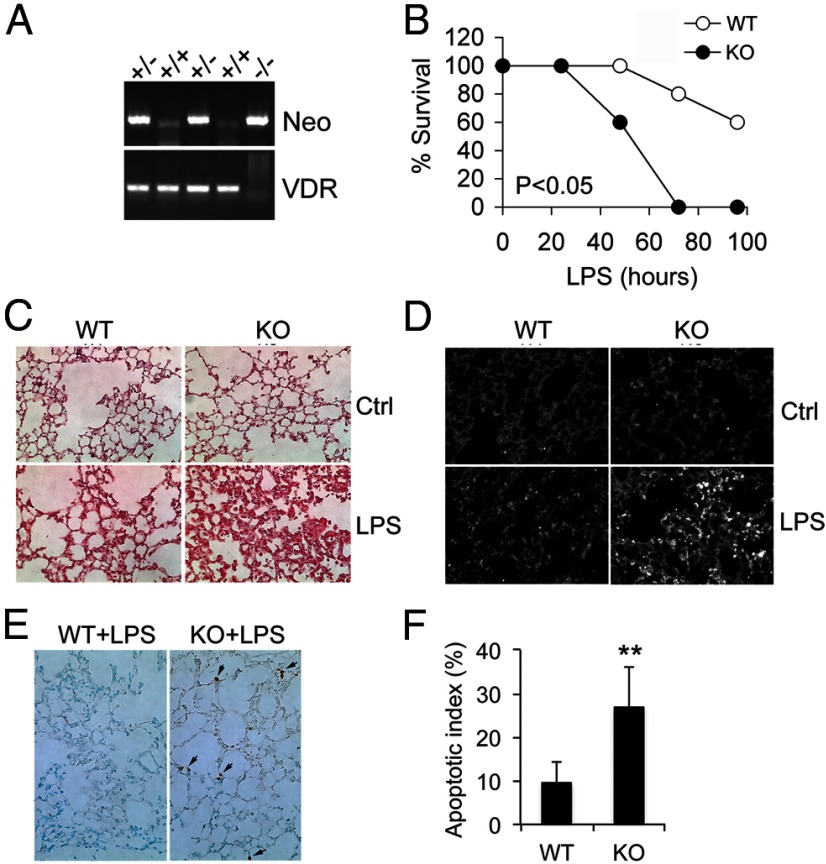
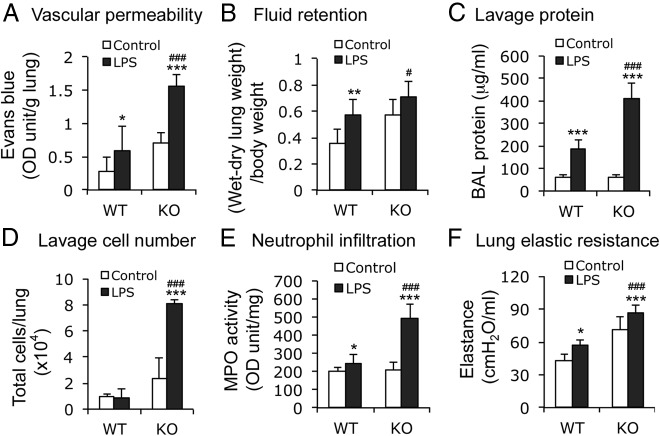
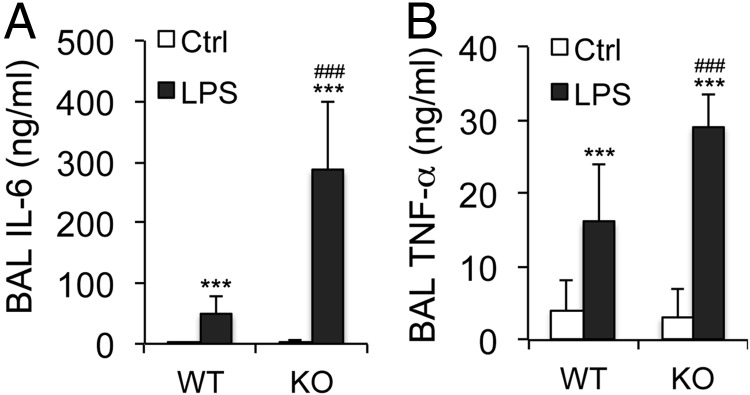
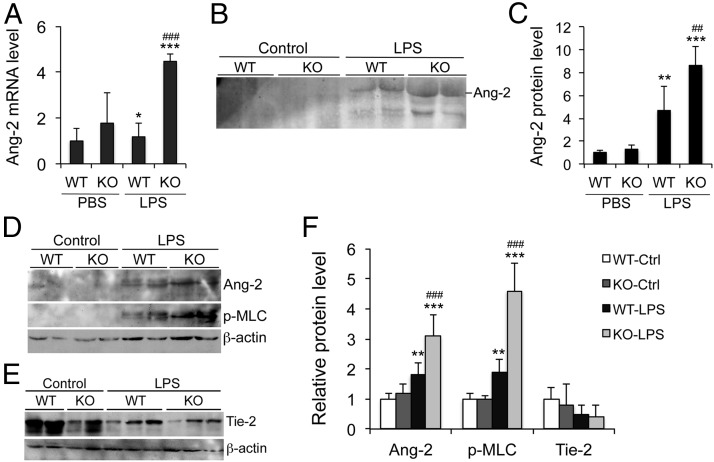
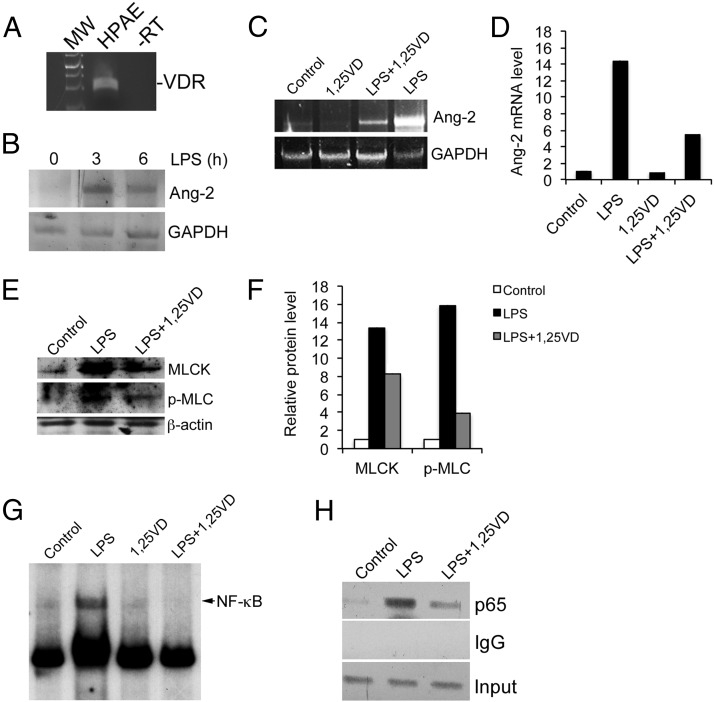
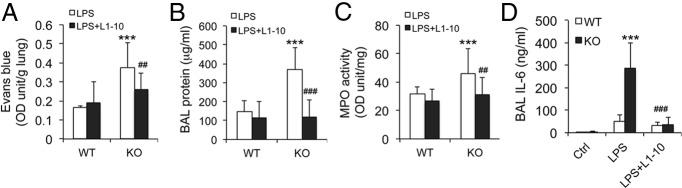
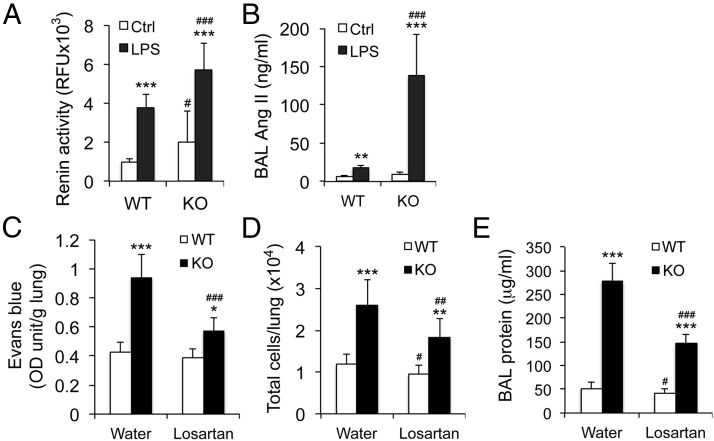
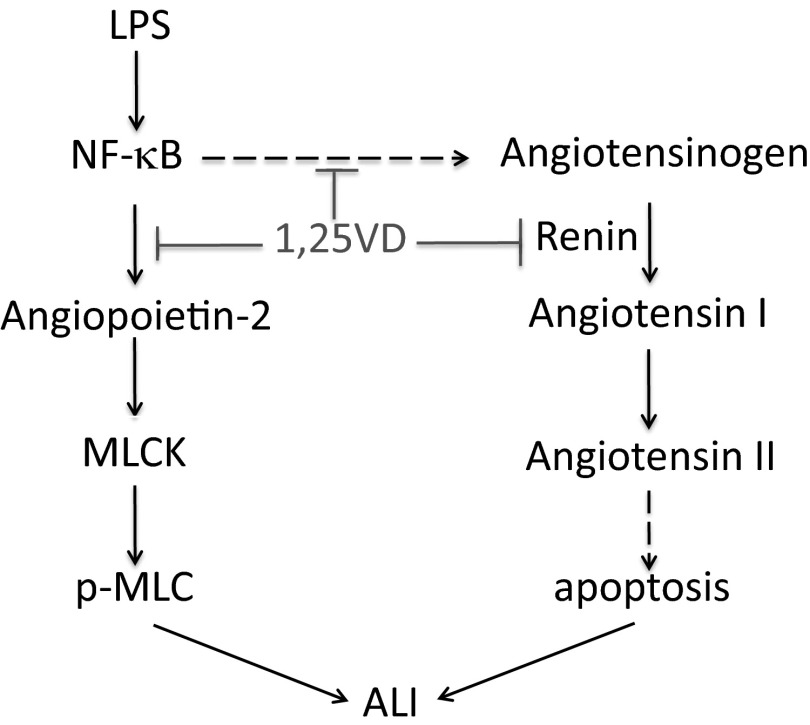
Acute lung injury (ALI) is a hallmark of systemic inflammation associated with high mortality. Although the vitamin D receptor (VDR) is highly expressed in the lung, its role in lung physiology remains unclear. We investigated the effect of VDR deletion on ALI using a lipopolysaccharide (LPS)-induced sepsis model. After LPS challenge VDR-null mice exhibited more severe ALI and higher mortality compared with wild-type (WT) counterparts, manifested by increased pulmonary vascular leakiness, pulmonary edema, apoptosis, neutrophil infiltration, and pulmonary inflammation, which was accompanied by excessive induction of angiopoietin (Ang)-2 and myosin light chain (MLC) phosphorylation in the lung. 1,25-Dihydroxyvitamin D blocked LPS-induced Ang-2 expression by blocking nuclear factor-κB activation in human pulmonary artery endothelial cells. The severity of lung injury seen in VDR-null mice was ameliorated by pretreatment with L1-10, an antagonist of Ang-2, suggesting that VDR signaling protects the pulmonary vascular barrier by targeting the Ang-2-Tie-2-MLC kinase cascade. Severe ALI in VDR-null mice was also accompanied by an increase in pulmonary renin and angiotensin II levels, and pretreatment of VDR-null mice with angiotensin II type 1 receptor blocker losartan partially ameliorated the severity of LPS-induced lung injury. Taken together, these observations provide evidence that the vitamin D-VDR signaling prevents lung injury by blocking the Ang-2-Tie-2-MLC kinase cascade and the renin-angiotensin system.
Figures
References
-
- Beck-Schimmer B, Schimmer RC, Pasch T. The airway compartment: chambers of secrets. News Physiol Sci. 2004;19:129–132 - PubMed
-
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334–1349 - PubMed
-
- Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol. 1995;163(3):510–522 - PubMed
-
- Tinsley JH, Teasdale NR, Yuan SY. Myosin light chain phosphorylation and pulmonary endothelial cell hyperpermeability in burns. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L841–L847 - PubMed
-
- Verin AD, Patterson CE, Day MA, Garcia JG. Regulation of endothelial cell gap formation and barrier function by myosin-associated phosphatase activities. Am J Physiol. 1995;269(1 Pt 1):L99–L108 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
